Abstract
Although widely studied, the natural diversity of the hard tick is not well known. In this study, we collected 194 sequences from 67 species, covering 7 genera of hard tick. The 5′ region of the mitochondrial cytochrome c oxidase subunit 1 region (586 bp) has been used to investigate intra- and inter-species variation and the phylogenetic tree of neighbor joining method has been used for assessment. As a result, by comparing the K2P-distance of intra- and interspecies, 30 samples (15.2%) shown that interspecies distance was larger than the minimum interspecfic distance. From the phylogenetic analysis, 86.8% (49) of the species were identified correctly at the genus level. On deeper analysis on these species suggested the possibility of presence cryptic species. Therefore, further work is required to delineate species boundaries and to develop a more complete understanding of hard tick diversity over larger scale.
-
Key words: CO1, hard tick, divergence, phylogenetic analyses
INTRODUCTION
Hard ticks are obligate ectoparasites, and seem to be second in importance only to mosquitoes as vectors of human and animal diseases [
1]. Tick-borne diseases cause a huge loss to the livestock industry and increase the risk of disease such as Lyme disease, babesiosis, human granulocytic ehrlichiosis, forest encephalitis, spotted fever, anaplasmosis, and Crimean–Congo hemorrhagic fever [
2–
4]. All species are exclusively hematophagous in all feeding stages. Hard ticks are distributed worldwide with their hosts range from wild to domestic vertebrates except fishes.
Traditionally, classifications and phylogenetic inferences for Ixodidae were based on morphological, biological and ecological characteristics, often suggesting host specificity as the main factor [
5,
6]. However, methods for species determination are limited when taxa are morphologically very similar, specimens are damaged, and in frequent cases where immature stages are not described or are engorged [
7].
Molecular systematics offered new possibilities to improve species recognition in hard ticks. ITS, 18S rDNA, 28S rDNA and other mitochondrial rDNA genes have been used to study these organisms and have played an important role in analyzing the classification and phylogenetics of hard ticks [
8–
10]. However, compared to the number of species of hard ticks, the extent of these studies are very limited [
11].
Until recently, there has been little effort to standardize the methods for molecular identification of hard ticks, and no one gene has been formally selected as an admitted DNA marker to deal with problems of classification and phylogenetics in hard ticks. So, we chose the mitochondrial cytochrome c oxidase subunit 1 (CO1) gene fragment as a candidate molecular marker, and collected 194 samples (from 67 species of 7 genera) of hard ticks. Intra- and interspecies genetic divergences were assessed using the Kimura 2-parameter (K2P) distance model. Phylogenetic tree were performed to analyse their relationship at evolutionary level.
MATERIALS AND METHODS
Sample collection
Ticks used in this study were collected from field sites and different hosts in various regions of China (
Table 1). After morphological identification, ticks were stored in 100% ethanol and conserved at 4°C. Only male and unfed adult specimens were used.
DNA was extracted from the ticks using a tissue kit (Qiagen AG, Basel, Switzerland) according to the manufacturer’s instructions. Each sample was cut with sterile scissors within a 1.5 ml microtube. After digestion with proteinase K (20 mg/ml), the samples were applied to the columns for DNA absorption and washing. Finally, the DNA was eluted in 100 ml of eluting buffer provided in the kit and stored at −20°C. The primers used for PCR were LCO1490 (5′-GGTCAACAAATCATAAAGATA-TTGG-3′) and HCO2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) [
12]. The 5′ region of
CO1 was amplified using the following thermal cycling program: 94°C for 5 min, 35 cycles at 94°C for 1 min, 53°C for 1 min, and 72°C for 1 min, followed by a final extension at 72°C for 8 min. PCR products were purified using a PCR purification kit (Takara, Shiga, Japan). Sequencing reactions were resolved on automated DNA sequencer.
Some CO1 sequences from the hard ticks were downloaded from GenBank. Sequences <500 bp in length, with ambiguous bases (more than 15 ‘Ns’), or those belonging to unnamed species (sequences with ‘sp.’ in the species name) were removed from the analysis. In addition, we checked all the sequences using BLAST analysis (E-value<0.001) to make sure that there were no host sequences in our data. The selected sequences were used to construct analysis datasets.
Sequence analysis
The
CO1 sequences were translated into amino acids with the program MEGA 4.0 in order to exclude sequencing errors and to avoid the inclusion of pseudogene sequences in the datasets. The sequences were aligned using ClustalW [
13], and genetic distances were computed using MEGA 4.0 according to the K2P distance model. The maximal/mean/minimum intra- and interspecies distances were used to represent species level divergence. Meanwhile, the maximal/mean/minimum intra-and intergenus distances were calculated to evaluate the genus level variation. Then a neighbor joining (NJ) tree was constructed and the genetic K2P distance was calculated within species and genera using MEGA 4.0. Evaluation of statistical confidence was based on 1,000 non-parametric bootstrap replicates. One soft tick isolate was used as the outgroup of phylogenetic tree.
RESULTS
Data acquisition
We collected 194 samples (36 from this study, 158 from GenBank) from 67 species and 7 genera of hard ticks (
Table 1 and
Supplementary Table S1). The mitochondrial
CO1 region of all samples collected in China was successfully amplified using PCR. The length of the PCR product was 707 bp. As some sequences of the
CO1 gene obtained from GenBank were shorter than 707 bp, all sequences were aligned with a consensus length of 586 bp, and no insertions, deletions, or stop codons were observed in any sequence. The sequences acquired in this study have been deposited in the GenBank database with accession numbers JQ737066-JQ737128.
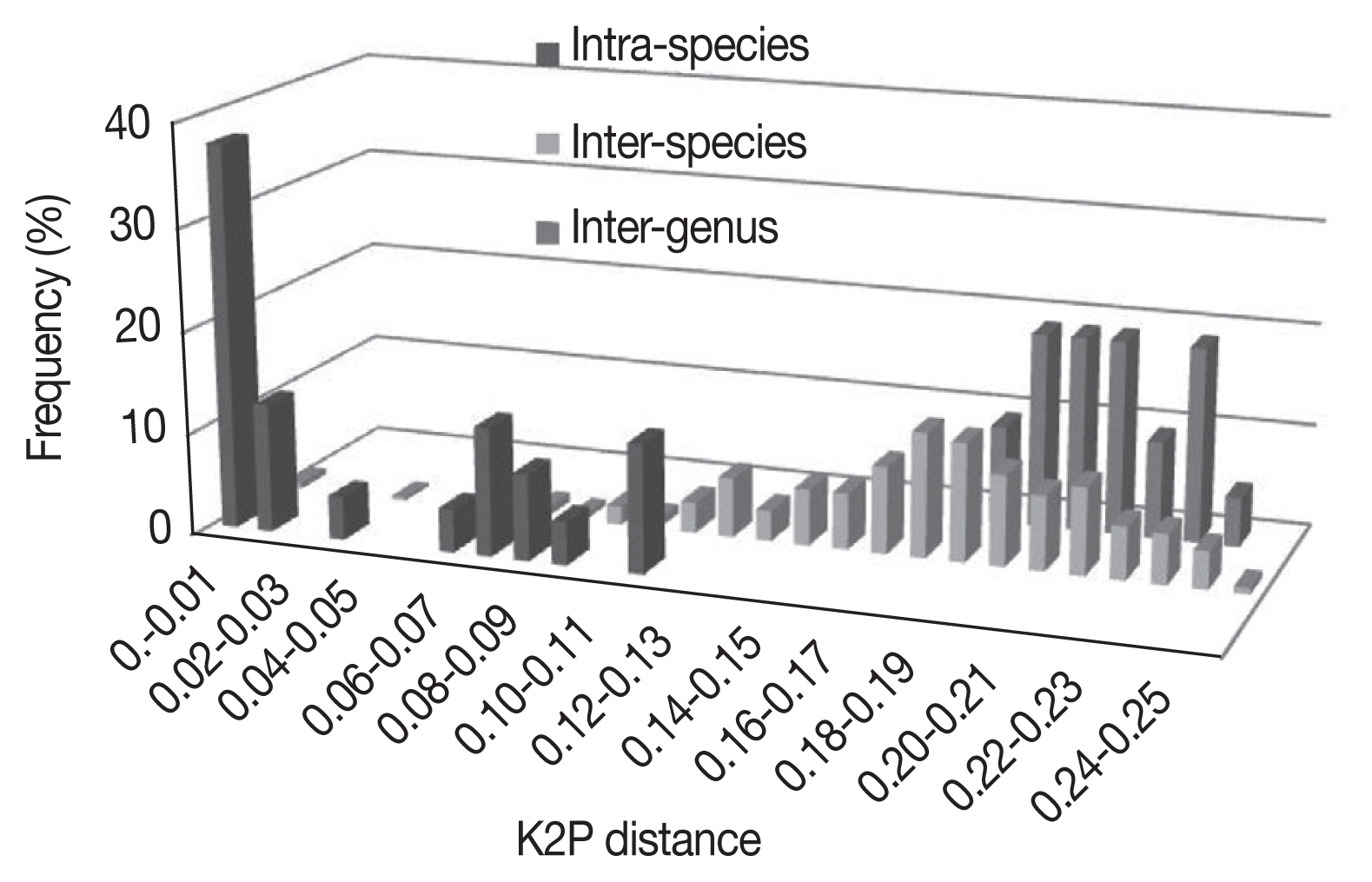
Using the K2P model, sample divergences at various taxonomic levels are shown in
Tables 2 and
3. As expected, the genetic divergence increased with higher taxonomic ranking: 0.001±0.001 to 0.016±0.003 at intraspecies level, 0.002±0.001 to 0.248±0.023 at interspecies level, 0.005±0.002 to 0.175±0.011 at intragenus level, and 0.186±0.012 to 0.243±0.016 at intergenus level. The
Bothriocroton showed the lowest mean intraspecies divergence (0.005±0.002), while
Rhipicephalus showed the highest mean intraspecies divergence (0.062±0.039) (
Fig. 1). The largest ratio between the average intra- and interspecies divergence was in the
Ixodes with a 7.5-fold difference, and the lowest ratio was in the
Dermacentor with a 2.4-fold difference. As shown in
Fig. 1, there was not a distinct gap between the distribution of the intra- and interspecies divergence. The overlapping regions were mainly distributed in the
R. turanicus, Hya. dromedarii, D. marginatus, D. silvarum, and
A. testudinarium.
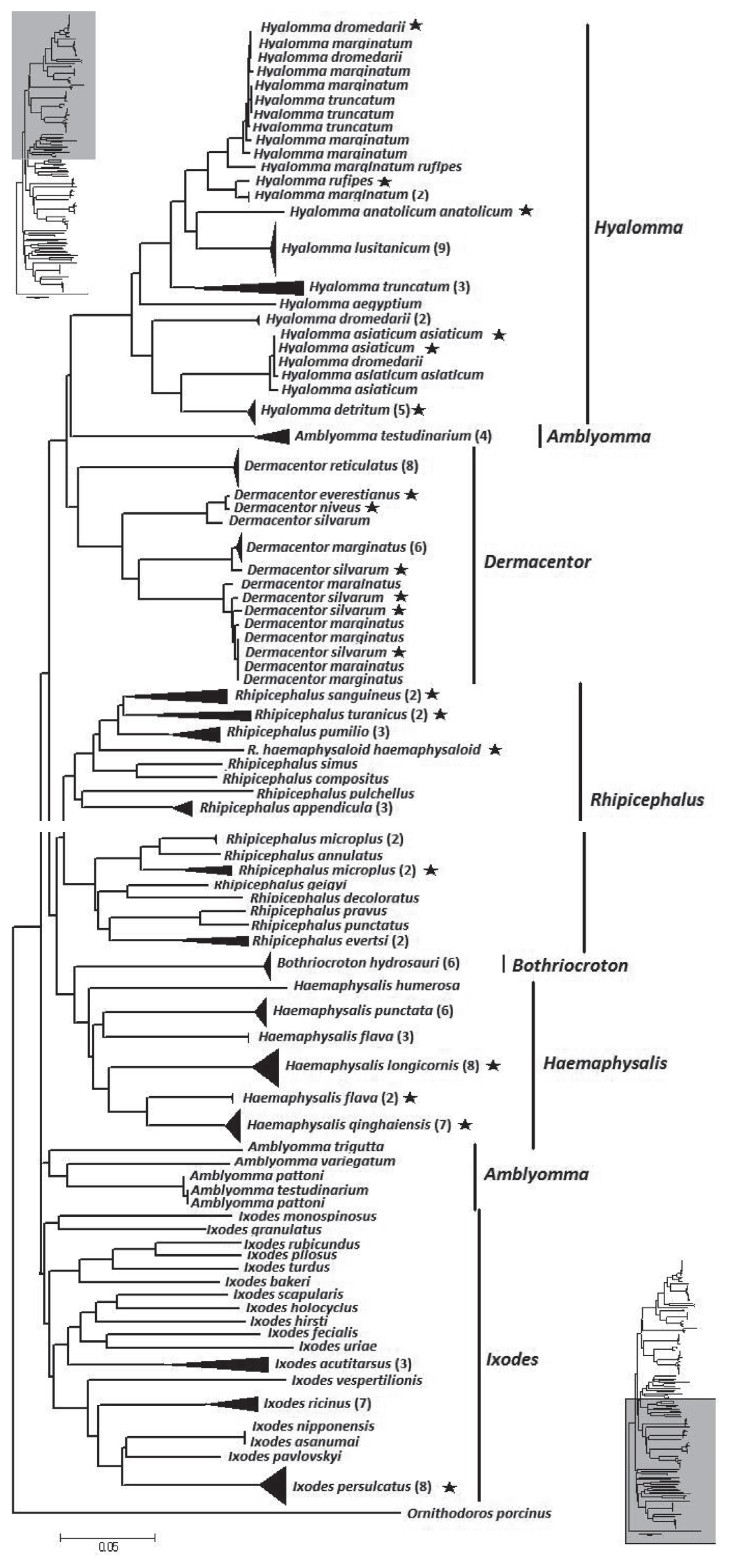
The NJ tree of the overall analysis is shown in
Fig. 2. The phylogenetic relationship at the genus level was well resolved with the exception of
Amblyomma. From the tree,
Hyalomma, Dermacentor, Amblyomma, and
Rhipicephalus formed 1 clade.
Bothriocroton and
Haemaphysalis formed another clade.
Ixodes as distinct difference at morphous to other hard ticks, formed a third clade. However, at a species level, 9 species (13.4%) did not form a monophyletic group. They were
Hya.
dromedarii, Hya. marginatum, Hya. asiaticum asiaticum, D. marginatus, D. silvarum, A. testudinarium, R. microplus, and
H. flava.
DISCUSSION
In this study, the mean sequence divergence in hard ticks (0.197±0.063) is higher than that observed in other organisms [
14–
16]. Such high values of genetic distance reflect possible biological diversity within the Ixodidae. Such as the distance between
Amblyomma testudinarium (HM193893) and
A. testudniarium (HM193895) was 0.112±0.010, and they were in different clades of the phylogenetic tree. However,
Rhipicephalus microplus and
Dermacentor marginatus also gave similar results. The reason may be geographic variation or comprise cryptic species [
17]. Additionally, the distance between the species
Dermacentor everestianus (JQ737079) and
D. niveus (JQ737080) was only 0.004±0.002, and also formed into 1 clade. Therefore, these analyses might indicate hybridization or a misidentification among these species.
The
CO1 gene appears to be an informative molecular marker on several taxonomic scales, but particularly at the species level [
18]. Our analysis shows a general increase in the molecular divergence of
CO1 with taxonomic rank. The diversity within species is especially high, with a maximum of 0.116±0.015. It makes
CO1 suitable for investigating intraspecies variation. DNA barcoding assumes that the genetic distances between species are greater than within species. In that way, clusters of similar sequences represent species, clearly separated from other clusters (species) [
19]. However, there also 30 samples where the maximum interspecies distance was larger than the minimum interspecfic distance. This means that the gap might be absent in these samples because of insufficient variation between them [
20,
21]. From the NJ phylogenetic tree, nine of the 67 species (13.4%) examined in this study (
Hya. dromedarii, Hya. marginatum, Hya. asiaticum asiaticum, Hya. truncatum, D. marginatus, D. silvarum, A. testudinarium, R. microplus, and
H. flava.) did not form a monophyletic group.
Hya. asiaticum asiaticum and
Hya. dromedarii shared similar morphologic characters from capitulum, scutum, Haller’s organ, peritrematal plate, the first caruncle, coax and spur of feet of adults and larval stages. Ecologically, these 2 species also share the same desert intertidal area. They are 2 different species proved by previous studies [
22–
24]. However, they formed one clade in this study. This phenomenon was also found for other hard ticks. For example,
Hya. dromedarii, Hya. marginatum and
Hya. truncatum formed a complex clade. These results agreed with some studies using mt 12S rDNA, 16S rDNA or ITS, in which
Hyalomma spp. shown high divergence distance and low bootstrap value [
25,
26]. As many results indicated that there is a high diversity in hard ticks [
27,
28].
This study provides that using the CO1 gene is a potential tool for species identification in Ixodidae. However, it is inadequate to use a single mitochondrial gene (CO1) for DNA taxonomy. Therefore, an integrative approach is needed to combine nuclear and mitochondrial genes, morphological characters, and ecological information into further studies of hard ticks.
Notes
-
CONFLICT OF INTEREST
All authors declare that they have no conflicts of interest.
Supplementary materials
Supplementary Table S1. The taxa and GenBank accession of 194 hard ticks used in this study
kjp-56-6-583-suppl.pdf
ACKNOWLEDGMENTS
This work was supported by the NSFC (No. 31560700), Provincial characteristic discipline open fund of Veterinary Medicine College, Gansu Agricultural University (GAU-XKJS-2018-08), Sheng Tongsheng innovation fund of Gansu Agricultural University (GSAU-STS-1422). The research was also facilitated by Specific Fund for Innovative Talent of Lanzhou City (No. 2014-2-11). We are indebted to international science editing for critical correction of this manuscript.
Fig. 1Frequency distribution of genetic K2P-distances in a 586 bp segment of the CO1 gene in Ixodidae at species and genus level.

Fig. 2Neighbor-joining tree of 194 isolates from the family Ixodidae and related species. The tree is constructed with 586 bp of CO1. Bracketed numbers represent the number of isolates sequenced for each species. Asterisk represent samples collected from China in this study.

Table 1Details of 36 samples collected from China in this study
Table 1
|
Genus |
Species |
Time |
Locality |
Source |
|
Hyalomma
|
Hya. dromedarii
|
Sep. 2010 |
Gansu |
Camel |
|
Hya. anatolicum anatolicum
|
Unknown |
Gansu |
Unknown |
|
Hya. detritum
|
Unknown |
Inner Mongolia |
Unknown |
|
Hya. asiaticum asiaticum
|
Unknown |
Inner Mongolia |
Ground |
|
Hya. asiaticum asiaticum
|
Jun. 2010 |
Xinjiang |
Cattle |
|
Hya. asiaticum
|
Jun. 2011 |
Gansu |
Camel |
|
Hya. rufipes
|
Jul. 2010 |
Gansu |
Goat |
|
|
Dermacentor
|
D. silvarum
|
Apr. 2010 |
Gansu |
Sheep |
|
D. silvarum
|
Apr. 2010 |
Gansu |
Goat |
|
D. silvarum
|
Apr. 2010 |
Gansu |
Sheep |
|
D. silvarum
|
May. 2011 |
Gansu |
Sheep |
|
D. silvarum
|
May. 2011 |
Gansu |
Sheep |
|
D. everestianus
|
May. 2011 |
Xizang |
Sheep |
|
D. niveus
|
Jun. 2011 |
Xizang |
Sheep |
|
|
Rhipicephalus
|
R. microplus
|
Jun. 2011 |
Gansu |
Cattle |
|
R. microplus
|
Jun. 2010 |
Guizhou |
Cattle |
|
R. sanguinens
|
May. 2010 |
Guangxi |
Dog |
|
R. haemaphysaloides haemaphysaloides
|
Jun. 2011 |
Sichuan |
Goat |
|
R. turanicus
|
May. 2010 |
Xinjiang |
Sheep |
|
|
Haemaphysalis
|
H. longicornis
|
May. 2011 |
Anhui |
Goat |
|
H. longicornis
|
Sep. 2010 |
Henan |
Sheep |
|
H. longicornis
|
Unknown |
Gansu |
Sheep |
|
H. longicornis
|
May. 2010 |
Hubei |
Sheep |
|
H. longicornis
|
Jun. 2011 |
Gansu |
Sheep |
|
H. longicornis
|
May. 2010 |
Zhejiang |
Sheep |
|
H. qinghaiensis
|
Apr. 2010 |
Gansu |
Sheep |
|
H. qinghaiensis
|
May. 2010 |
Gansu |
Sheep |
|
H. qinghaiensis
|
May. 2011 |
Gansu |
Sheep |
|
H. qinghaiensis
|
Jun. 2011 |
Qinghai |
Ground |
|
H. qinghaiensis
|
Jun. 2011 |
Qinghai |
Sheep |
|
H. qinghaiensis
|
May. 2008 |
Gansu |
Ground |
|
H. flava
|
Sep. 2010 |
Henan |
Sheep |
|
|
Ixodes
|
I. persulcatus
|
Jun. 2011 |
Xinjiang |
Sheep |
Table 2Measures of inter- and intra-species divergences for CO1 sampled in 7 genera of Ixodidae
Table 2
|
Intra-species distance |
Inter-species distance |
|
|
|
Minimum |
Mean |
Maximum |
Minimum |
Mean |
Maximum |
|
Hyalomma
|
0.004±0.002 |
0.039±0.046 |
0.110±0.010 |
0.035±0.006 |
0.113±0.027 |
0.155±0.017 |
|
|
Dermacentor
|
0.003±0.001 |
0.050±0.042 |
0.084±0.008 |
0.002±0.001 |
0.122±0.058 |
0.179±0.016 |
|
|
Haemaphysalis
|
0.008±0.002 |
0.033±0.042 |
0.016±0.003 |
0.150±0.016 |
0.175±0.021 |
0.191±0.019 |
|
|
Bothriocroton
|
0.005±0.002 |
0.005±0.002 |
0.005±0.002 |
0.000±0.000 |
0.000±0.000 |
0.000±0.000 |
|
|
Rhipicephalus
|
0.014±0.004 |
0.062±0.039 |
0.116±0.015 |
0.051±0.010 |
0.156±0.028 |
0.207±0.020 |
|
|
Amblyomma
|
0.002±0.002 |
0.057±0.077 |
0.112±0.010 |
0.147±0.016 |
0.177±0.028 |
0.206±0.018 |
|
|
Ixodes
|
0.001±0.001 |
0.026±0.043 |
0.077±0.010 |
0.094±0.017 |
0.196±0.030 |
0.248±0.023 |
Table 3Measures of inter- and intragenus divergences for CO1 sampled in family Ixodidae
Table 3
|
Intra-genus distance |
Inter-genus distance |
|
|
|
Minimum |
Mean |
Maximum |
Minimum |
Mean |
Maximum |
|
Ixodidae |
0.005±0.002 |
0.118±0.056 |
0.175±0.011 |
0.186±0.012 |
0.211±0.017 |
0.243±0.016 |
References
- 1. Jongejan F, Uilenberg G. The global importance of ticks. Parasitology 2004;129(suppl):3-14.
- 2. Banumathi B, Vaseeharan B, Rajasekar P, Prabhu NM, Ramasamy P, Murugan K, Canale A, Benelli G. Exploitation of chemical, herbal and nanoformulated acaricides to control the cattle tick, Rhipicephalus (Boophilus) microplus - A review. Vet Parasitol 2017;244:102-110.
- 3. Chen Z. Taxonomic and Systematic Research of Chinese Ticks and Biological Characteristic Analysis of Two Hard Tick Species. Shijiazhuang, China. Hebei Normal University; 2010.
- 4. Estrada-Peña A, Ayllón N, de la Fuente J. Impact of climate trends on tick-borne pathogen transmission. Front Physiol 2012;3:64.
- 5. McCoy KD, Léger E, Dietrich M. Host specialization in ticks and transmission of tick-borne diseases: a review. Front Cell Infect Microbiol 2013;3:57.
- 6. Klompen JS, Black WC 4th, Keirans JE, Oliver JH Jr. Evolution of ticks. Annu Rev Entomol 1996;41:141-161.
- 7. Nava S, Guglielmone AA, Mangold AJ. An overview of systematics and evolution of ticks. Front Biosci 2009;14:2857-2877.
- 8. Li HY, Zhao SS, Hornok S, Farkas R, Guo LP, Chen CF, Shao RF, Lv JZ, Wang YZ. Morphological and molecular divergence of Rhipicephalus turanicus tick from Albania and China. Exp Appl Acarol 2017;73:493-499.
- 9. Livanova NN, Tikunov AY, Kurilshikov AM, Livanov SG, Fomenko NV, Taranenko DE, Kvashnina AE, Tikunova NV. Genetic diversity of Ixodes pavlovskyi and I. persulcatus (Acari: Ixodidae) from the sympatric zone in the south of Western Siberia and Kazakhstan. Exp Appl Acarol 2015;67:441-456.
- 10. Marrelli MT, Souza LF, Marques RC, Labruna MB, Matioli SR, Tonon AP, Ribolla PE, Marinotti O, Schumaker TT. Taxonomic and phylogenetic relationships between neotropical species of ticks from genus Amblyomma (Acari: Ixodidae) inferred from second internal transcribed spacer sequences of rDNA. J Med Entomol 2007;44:222-228.
- 11. Chitimia L, Lin RQ, Cosoroaba I, Wu XY, Song HQ, Yuan ZG, Zhu XQ. Genetic characterization of ticks from southwestern Romania by sequences of mitochondrial cox1 and nad5 genes. Exp Appl Acarol 2010;52:305-311.
- 12. Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 1994;3:294-299.
- 13. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994;22:4673-4680.
- 14. Matzen da Silva J, Creer S, dos Santos A, Costa AC, Cunha MR, Costa FO, Carvalho GR. Systematic and Evolutionary Insights Derived from mtDNA CO1 Barcode Diversity in the Decapoda (Crustacea: Malacostraca). PLoS One 2011;6:e19449.
- 15. Kerr KC, Stoeckle MY, Dove CJ, Weigt LA, Francis CM, Hebert PD. Comprehensive DNA barcode coverage of North American birds. Mol Ecol Notes 2007;7:535-543.
- 16. Ros VI, Breeuwer JA. Spider mite (Acari: Tetranychidae) mitochondrial CO1 phylogeny reviewed: host plant relationships, phylogeography, reproductive parasites and barcoding. Exp Appl Acarol 2007;42:239-262.
- 17. Rees DJ, Dioli M, Kirkendall LR. Molecules and morphology: evidence for cryptic hybridization in African Hyalomma (Acari: Ixodidae). Mol Phylogenet Evol 2003;27:131-142.
- 18. Waugh J. DNA barcoding in animal species: progress, potential and pitfalls. Bioessays 2007;29:188-197.
- 19. Hebert PD, Ratnasingham S, deWaard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci 2003;270(suppl):96-99.
- 20. Sheth BP, Thaker VS. DNA barcoding and traditional taxonomy: an integrated approach for biodiversity conservation. Genome 2017;60:618-628.
- 21. Meyer CP, Paulay G. DNA barcoding: error rates based on comprehensive sampling. PLoS Biol 2005;3:e422.
- 22. Apanaskevich DA, Horak IG. The genus Hyalomma. XI. Redescription of all parasitic stages of H. (Euhyalomma) asiaticum (Acari: Ixodidae) and notes on its biology. Exp Appl Acarol 2010;52:207-220.
- 23. Apanaskevich DA, Schuster AL, Horak IG. The genus Hyalomma: VII. Redescription of all parasitic stages of H. (Euhyalomma) dromedarii and H. (E.) schulzei (Acari: Ixodidae). J Med Entomol 2008;45:817-831.
- 24. Chen Z, Yu Z, Yang X, Zheng H, Liu J. The life cycle of Hyalomma asiaticum kozlovi Olenev, 1931 (Acari: Ixodidae) under laboratory conditions. Vet Parasitol 2009;160:134-137.
- 25. Kaur H, Chhilar JS, Chhillar S. Mitochondrial 16S rDNA based analysis of some hard ticks belonging to genus Hyalomma Koch, 1844 (Acari: Ixodidae). J Adv Parasitol 2016;3:32-48.
- 26. Hekimoglu O, Ozer AN. Distribution and phylogeny of Hyalomma ticks (Acari: Ixodidae) in Turkey. Exp Appl Acarol 2017;73:501-519.
- 27. Bursali A, Keskin A, Tekin S. A review of the ticks (Acari: Ixodida) of Turkey: species diversity, hosts and geographical distribution. Exp Appl Acarol 2012;57:91-104.
- 28. Shemshad K, Rafinejad J, Kamali K, Piazak N, Sedaghat MM, Shemshad M, Biglarian A, Nourolahi F, Valad Beigi E, Enayati AA. Species diversity and geographic distribution of hard ticks (Acari: Ixodoidea: Ixodidae) infesting domestic ruminants, in Qazvin Province, Iran. Parasitol Res 2012;110:373-380.