Abstract
Echinostoma revolutum is a zoonotic food-borne intestinal trematode that can cause intestinal bleeding, enteritis, and diarrhea in human and birds. To identify a suspected E. revolutum trematode from a red-crowned crane (Grus japonensis) and to reveal the genetic characteristics of its mitochondrial (mt) genome, the internal transcribed spacer (ITS) and complete mt genome sequence of this trematode were amplified. The results identified the trematode as E. revolutum. Its entire mt genome sequence was 15,714 bp in length, including 12 protein-coding genes, 22 transfer RNA genes, 2 ribosomal RNA genes and one non-coding region (NCR), with 61.73% A+T base content and a significant AT preference. The length of the 22 tRNA genes ranged from 59 bp to 70 bp, and their secondary structure showed the typical cloverleaf and D-loop structure. The length of the large subunit of rRNA (rrnL) and the small subunit of rRNA (rrnS) gene was 1,011 bp and 742 bp, respectively. Phylogenetic trees showed that E. revolutum and E. miyagawai clustered together, belonging to Echinostomatidae with Hypoderaeum conoideum. This study may enrich the mitochondrial gene database of Echinostoma trematodes and provide valuable data for studying the molecular identification and phylogeny of some digenean trematodes.
-
Key words: Echinostoma revolutum, red-crowned crane, mitochondrial genome, PCR
Echinostomes, family Echinostomatidae, are intestinal trematodes, which infect birds and mammals, including humans, causing echinostomiasis characterized by mucosal ulceration and bleeding in heavy infection [
1]. Currently, 21 zoonotic
echinostoma species, belonging to 9 genera, have been recorded [
2]. Among them,
Echinostoma is the most important genus. Seven zoonotic
Echinostoma species are documented:
E. revolutum, E. hortense, E. angustitestis, E. echinatum, E. cinetorchis, E. ilocanum, and
E. macrorchis. E. revolutum (Froelich, 1802) Looss, 1899 was the first described echinostome species [
1]. It was originally identified as an intestinal fluke of ducks but now is well-established to inhabit the rectum and cecum of several avian and mammalian species, including geese, muskrats, and humans [
1,
3].
E. revolutum suckers, head spines, and body spines can cause mechanical damage to the intestinal wall of the host, leading to intestinal bleeding, enteritis, and diarrhea. Besides, this trematode can also absorb large amounts of nutrients, leading to wasting and stunted growth in sick birds.
E. revolutum is widely distributed in the world, including Asia, Europe, Africa, Australia, New Zealand, and the Americas [
1]. In Asia, human infection with
E. revolutum has been reported in China, Indonesia, and Thailand [
4]. The main transmission route of human infection is the consumption of raw or uncooked aquatic animals, such as snails and frogs [
1].
The morphological classification and identification of the echinostome species have been intensively studied for a long time due to the large number of Echinostomatidae family members [
5]. According to the morphological characteristics of these flukes, Chai [
1] formulated a key for the identification of common animal and human echinostomes. With the development of molecular biology techniques, especially the emergence of polymerase chain reaction (PCR) and sequencing technology, the species identification of parasites has entered the era of DNA analysis. Molecular markers can improve our understanding of phylogeny and genetic evolution of parasites [
6]. The internal transcribed spacers (ITS1, ITS2) of ribosomal DNA (rDNA) are often used as molecular markers to identify and classify parasite species [
7]. Presently, Saijuntha et al. [
8] has reported the intraspecific variation of ITS1 sequences of
E. revolutum in different regions of Thailand and Laos.
Mitochondrial (mt) genome (mtDNA) is a unique genetic system independent from the nuclear genome, which is characterized by small molecular weight, conservative and relatively stable structure, maternal inheritability, low recombination and high mutation [
9]. Therefore, it is widely used to explore population relationship, genetic evolution, and comparative genomics [
10]. With the continuous development of bioinformatics and sequencing technology, more and more mtDNA of trematodes has been decoded [
11], and their structural characteristics have been gradually analyzed. The complete mitochondrial genome of
Ogmocotyle sikae and
Clonorchis sinensis has been sequenced and systemically analyzed [
12,
13]. Among nearly 500 species in family Echinostomatidae, the mitochondrial genome of only 2 species,
Hypoderaeum conoideum (KM111525) and
Echinostoma hortense (NC028010) have been reported in the literature [
14]. However, the mitochondrial genome of
E. revolutum has not been identified yet.
This study aimed to identify E. revolutum isolated from a red-crowned crane (Grus japonensis) based on ITS sequence-PCR amplification, to describe for the first time the complete mtDNA sequence of this parasite and to elucidate its phylogenetic relationship with other digenean trematodes.
Twenty-seven flukes were collected from an albendazole-treated red-crowned crane (Grus japonensis) at the Star-Lake Ecological Park in Zhaoqing city, Guangdong Province. The trematode specimens were washed with distilled water, fixed in 70% alcohol, and stored at −20°C until further use. These trematodes were identified as Echinosoma species based on its morphological characteristics. Individual echinostome specimens were selected and washed with sterilized double-distilled water (ddH2O) for 5 times. Then, the total genomic DNA was extracted from the individual specimens according to the manufacturer’s instructions of Wizad® SV Genomic DNA Purification System (Promega, Madison, Wisconsin, USA) and stored at −20°C for later use.
To amplify ITS sequence of the isolated flukes, a forward primer (ERF: 5′-AGGAGACTTGGGCTACGGCT-3′) and reverse primer (ERR: 5′-CGATAGAGTGACAGTAACGGAACG-3′) were designed based on the ITS sequence of echinostomes published on NCBI. The expected fragment is 713 bp. PCR was performed referring to Shi et al. [
15]. PCR products were examined by 1.5% agarose gel electrophoresis, re-harvested using the OMEGA Gel Extraction Kit (Omega Bio-Tek, Norcross, Georgia, USA) and sent to Sangon Biotech (Shanghai) company for sequencing. Our sample ITS sequence was used to search GenBank using BLAST to determine its homology with other known sequences. The phylogenetic tree was constructed using the neighbor-joining method by MEGA 5 software.
Six pairs of primers were designed using Primer Premier 5 software based on the conserved regions in the mt genome sequence of
E. hortense (GenBank: NC028010) and
E. caproni (GenBank: AP017706). All the primers were synthesized by Sangon Biotech (Shanghai) company. These primers were used to amplify the complete mt genome in 6 overlapping fragments from a total genomic DNA sample of 1
E. revolutum individual. Long PCR amplification was performed referring to Zhang et al. [
16]. PCR products were examined by 1.5% agarose gel electrophoresis. The electrophoresis product was extracted according to the manufacturer’s instructions of the OMEGA Gel Extraction Kit (Omega Bio-Tek). After ligating the purified product with PMD18-T cloning vector (TaKaRa, Dalian, China), the target fragment was transferred into
Escherichia coli DH5α cells (TaKaRa). Then, the plasmids were extracted from PCR-positive clones following the manufacturer’s protocol of the OMEGA plasmid DNA Mini Kit (Omega Bio-Tek), and sent to Sangon Biotech (Shanghai, China) company for sequencing.
The high-quality sequences were detected by BioEdit version 7.0. The sequences of the 6 genomic fragments were manually assembled into a complete mt genome by removing overlapping sequences with the SeqMan module of DNASTAR software version 5.0 (DNASTAR Inc., Madison, Wisconsin, USA). The assembled mt genome was further aligned against the published mt genome of related trematodes using the online software (
http://dogma.ccbb.utexas.edu/) combined with the MegAlign module of DNASTAR software version 5.0 (DNASTAR) to determine its composition, boundaries between genes, start codons, and stop codons. The AT content was calculated using the EditSeq module of DNASTAR software version 5.0 (DNASTAR). Twenty-two tRNA genes of
E. revolutum were identified and their secondary structures were predicted using online tRNA scan program (
http://lowelab.ucsc.edu/tRNAscan-SE/) combined with manual proofreading.
The concatenated amino acid sequences of
E. revolutum mt genome, conceptually translated from the 12 protein-coding genes, were aligned with those of the published mt genomes from 19 selected digenean trematodes. The sequence of the nematode parasite,
Ascaris suum (NC_001327) was included as outgroup. Three different inference methods, namely the maximum likelihood, maximum parsimony and neighbor-joining, were used for phylogenetic analyses. The ML method was conducted using PhyML 3.0 [
17] based on the Akaike information criterion, branch supports were estimated by bootstrap analysis of 1,000 replicates. The MP method in PAUP* 4.0b10 [
18] with default parameters and the bootstrap analyses (with 1,000 replicates) based on Branch and Bound algorithm. The NJ method in MEGA 5.0 [
19]. Phylograms were drawn using FigTree v.1.42 software [
20].
Adult worms were long leaf-shaped, 9–12 mm long and 1.4–1.6 mm wide. Microscopically, 37 cephalic spines were observed, and the 2 testicles were oval and tandemly arranged. The amplified fragment was 713 bp in length, consisted of partial ITS1 (<196 bp), complete 5.8S (196–356 bp) and partial ITS2 (357–713 bp) fragment (GenBank MN874065). Its sequence homology to the previously published sequence of
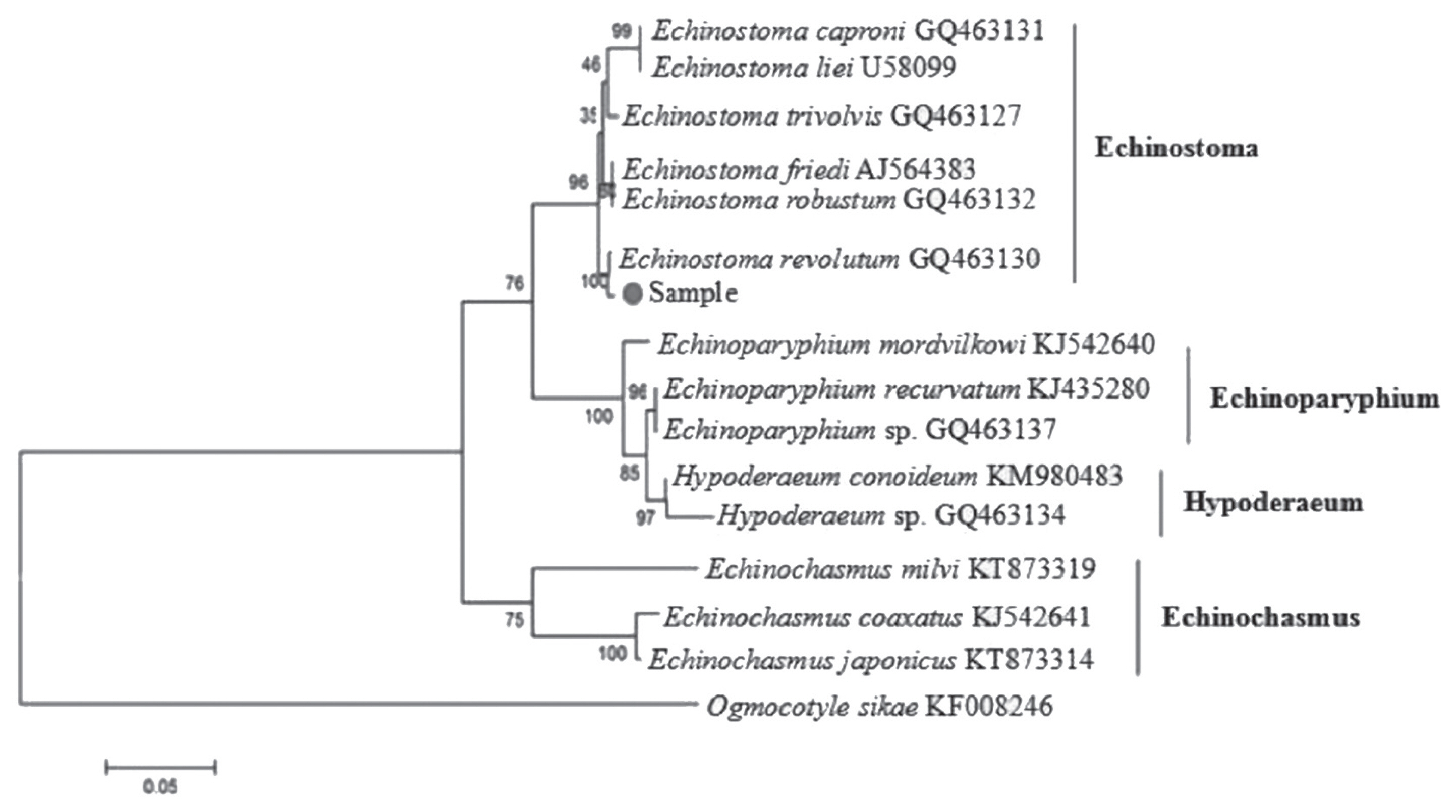
E. revolutum (GQ463130) was 99.00%. The phylogenetic tree (
Fig. 1) showed that our sample was clustered into the same branch with
E. revolutum (GQ463130). Thus, it was confirmed that the fluke isolated from the red-crowned crane is
E. revolutum.
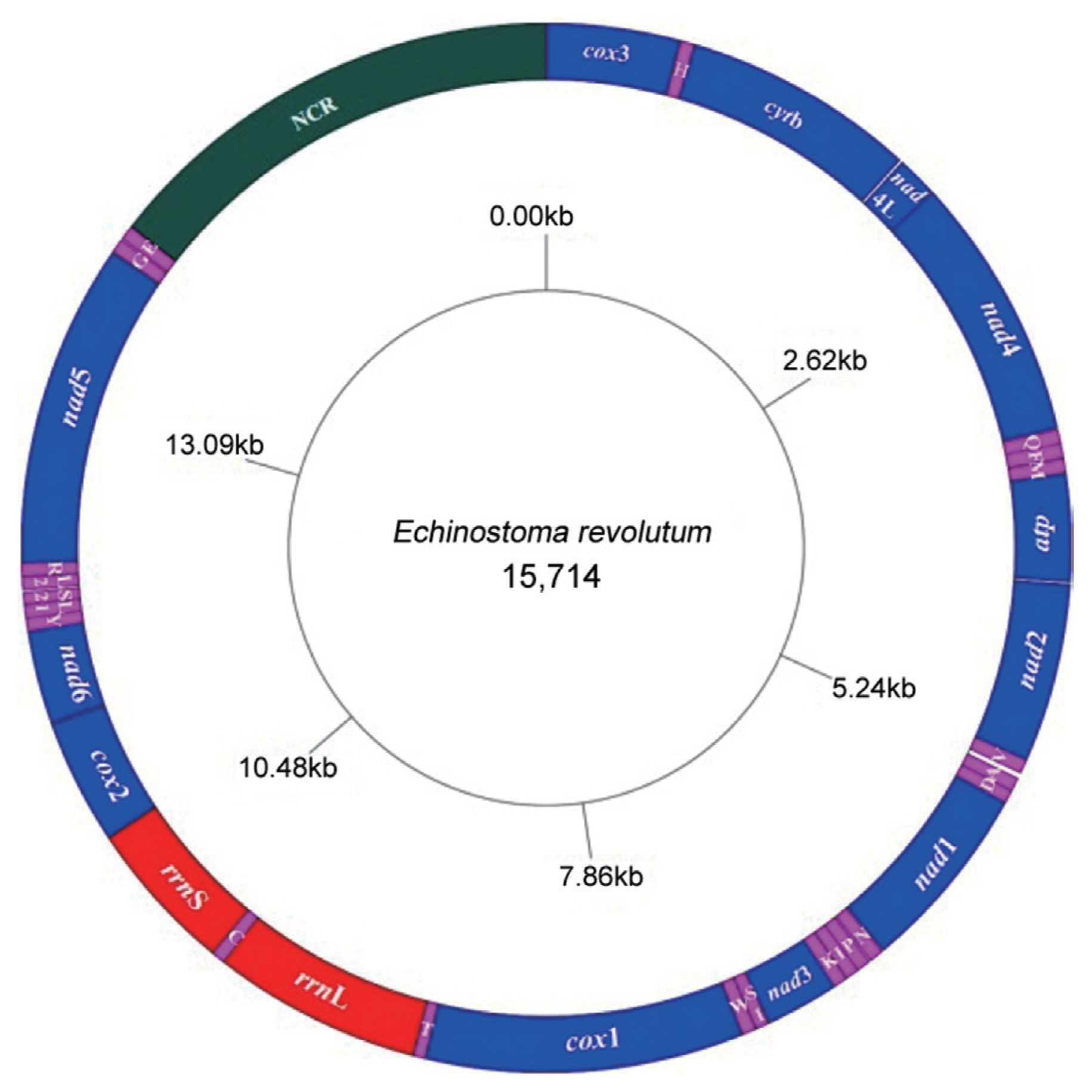
The complete mitochondrial genome sequence (GenBank: MN116706) of
E. revolutum was 15,714 bp in length and comprised 36 genes, consisted of 12 protein-coding genes (
cox1–3,
nad1–6,
nad4L,
atp6, and
cytb), 22 tRNA genes, and 2 ribosomal RNA genes (
rrnL and
rrnS), and a single non-coding region (NCR) (
Fig. 2). All genes were transcribed in the same direction. The arrangements and length of all mt genes are shown in
Table 1. The mitochondrial genes were tightly arranged with 25 intergenic spacers throughout the mitochondrial genome, and short intervals ranging from 1 bp to 29 bp in length. There was only 38 bp overlap between
nad4L and
nad4 genes. The nucleotide compositions were as follows: A=19.02%, G=27.34%, T=42.71%, C=10.93%, A+T=61.73%, and G+C=38.27%, showing obvious preference for AT.
All the 12 protein-coding genes (PCGs) (
Table 1) in the mitochondrial genome of
E. revolutum showed obvious AT preference, among which
nad4L gene had the highest AT content (66.26%) and
atp6 gene had the lowest AT content (59.73%). The 12 protein-coding genes used ATG (50.00%) or GTG (50.00%) as start codons. Concerning stop codons, 8 PCGs (67.00%) used TAG and 4 PCGs (33.00%) used TAA, without TA or T incomplete stop codons. AT-skew values of all protein-coding genes were less than zero, and GC-skew values were higher than zero.
There were 22 tRNA genes in the mitochondrial genome of E. revolutum, ranging in length from 57 to 70 bp. Except for the D-loop structure of tRNA-SerGCU, all the other tRNAs showed the typical cloverleaf structure. The amino acid acceptor arms of all tRNA secondary structures were composed of 7 bp, and the anti-codon region consisted of a stem with 5 bp and a ring with 7 bases. The tRNA-SerGCU was the shortest one among the 22 tRNAs, containing a complete TΨC arm, while the DHU loop was replaced by 7 bases.
The large subunit 16S (rrnL) was 1,011 bp long, located between tRNA-Thr and tRNA-Cys genes, without gene interval, and with the AT content of 60.73%. The small subunit 12S (rrnS) was 742 bp long, located between the tRNA-Cys and cox2 genes, without gene interval, and with AT content of 58.28%. There was a single non-coding region (NCR) in the mitochondrial genome of E. revolutum, located between tRNA-Glu and cox3, with 2,280 bp length and 58.86% AT content. Compared to the 12 protein-coding genes, the NCR had the lowest AT content.
Phylogenetic relationships of E. revolutum with the selected 19 digenean trematodes using 3 analytical methods (MP, ML, and NJ) were identical in their topology. The phylogenetic trees constructed based on the 12 protein-coding genes showed that E. revolutum and E. miyagawai clustered together and belonged to the branch of Echinostomatidae with Hypoderaeum conoideum. In Digenea, Echinostomatidae and Fasciolidae clustered together; Notocotylidae and Paramphistomatidae together; Paragonimidae, Opisthorchiidae, and Heterophyidae together; and Clinostomatidae was in a separate branch. Echinostomatidae and Fasciolidae were relatively closer to each other than to other families in Digenea, while E. revolutum and E. miyagawai were the closest species in Echinostomatidae with high nodal support.
In this experiment, we described the molecular identification of E. revolutum based on the primers designed according to the ITS sequence of echinostomes available in GenBank. These trematode samples were isolated from a red-crowned crane (Grus japonensis) medicated with albendazole anthelmintic drug. Grus japonensis is one of the rare birds on the verge of extinction in the world and has been listed as one of China’s key protected birds. This bird mainly feeds on snails, freshwater fish and shrimp, making it more vulnerable to infection with a variety of trematode parasites, including E. revolutum. The PCR amplified fragment was 713 bp in length, with 99.00% homology to E. revolutum (GQ463130). The phylogenetic tree constructed by the NJ method showed that our sample was clustered in the same group with E. revolutum (GQ 463130), which confirms the fluke species as E. revolutum.
Our results demonstrated that
E. revolutum mt genome composition was the same as that of
E. miyagawai (NC039532) and
E. hortense (KR062182) [
21,
22]. However, another member of the family Echinostomatidae (
Echinochasmus japonicas) has 2 non-coding regions (LNR and SNR) separated by
trnE [
23]. This result suggests that there are differences in the composition of the mitochondrial genome of Echinostomatidae trematodes. The size of the mitochondrial genome largely depends on non-coding regions. For example,
Clonorchis sinensis has a long non-coding region [
24], so its mitochondrial genome is a longer one. Meanwhile, 2 non-coding regions of
Schistosoma spindale are short [
25], so its mitochondrial genome is a shorter one. Non-coding and spacer regions mainly control mitochondrial transcription and translation processes through repeat sequences and some special structures and microsatellite structures [
26]. Therefore, they have small evolutionary pressure, but their lengths change greatly. The mitochondrial genome of
E. revolutum had a tight structure. The 36 genes were separated by short gene intervals, and there was only 38 bp overlap between
nad4L and
nad4 genes, which was consistent with the characteristics of the mitochondrial genome of Digenea trematodes [
26].
E. revolutum mt DNA nucleotide composition had obvious AT preference (61.73%). This finding was consistent with the mitochondrial genome of most trematodes, where the AT content in the majority of trematodes mt genome is within the range (60.00–70.00%), except for
Paragonimus westermani, with 51.50% AT content [
22]. There are no introns in the 12 protein-coding genes in the whole mitochondrial genome of
E. revolutum, with ATG and GTG as the start codons, TAG and TAA as stop codons, and no incomplete stop codon. By contrast, incomplete stop codons are occasionally used in other trematodes, such as
nad3 of
Gyrodactylus derjavinoides and
nad2 of
Gyrodactylus thymalli using T as the terminating codon for translation. Incomplete codons in the mitochondrial genome can also serve as a signal for transcription termination because base A can be inserted after T or TA during the post-transcriptional processing to complete the termination codon [
27]. All the 12 protein-coding genes in the complete mitochondrial genome of
E. revolutum have obvious AT preference, and the higher AT preference makes the gene structure more stable and reduces the probability of gene mutation.
In summary, the molecular identification and whole mitochondrial genome sequence analysis of E. revolutum from Grus japonensis were conducted in this paper, which not only enriched the mitochondrial gene database of E. revolutum, but also provided important information for studying the identification, and phylogenetic relations among some trematodes in order Digenea.
Notes
-
The authors declare no conflict of interest related to this study.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Natural Science Foundation of China (Grant No. 31672541) and the Science and Technology Planning Project of Guangdong Province, China (Grant No. 2014A020214005). The authors would like to thank Mr. Wu Chen, the personnel of Guangzhou Zoo, for help in collection of the samples.
Fig. 1Phylogenetic tree of Echinostoma revolutum and other Echinostomatidae trematodes constructed on ITS sequence using neighbor-joining method.
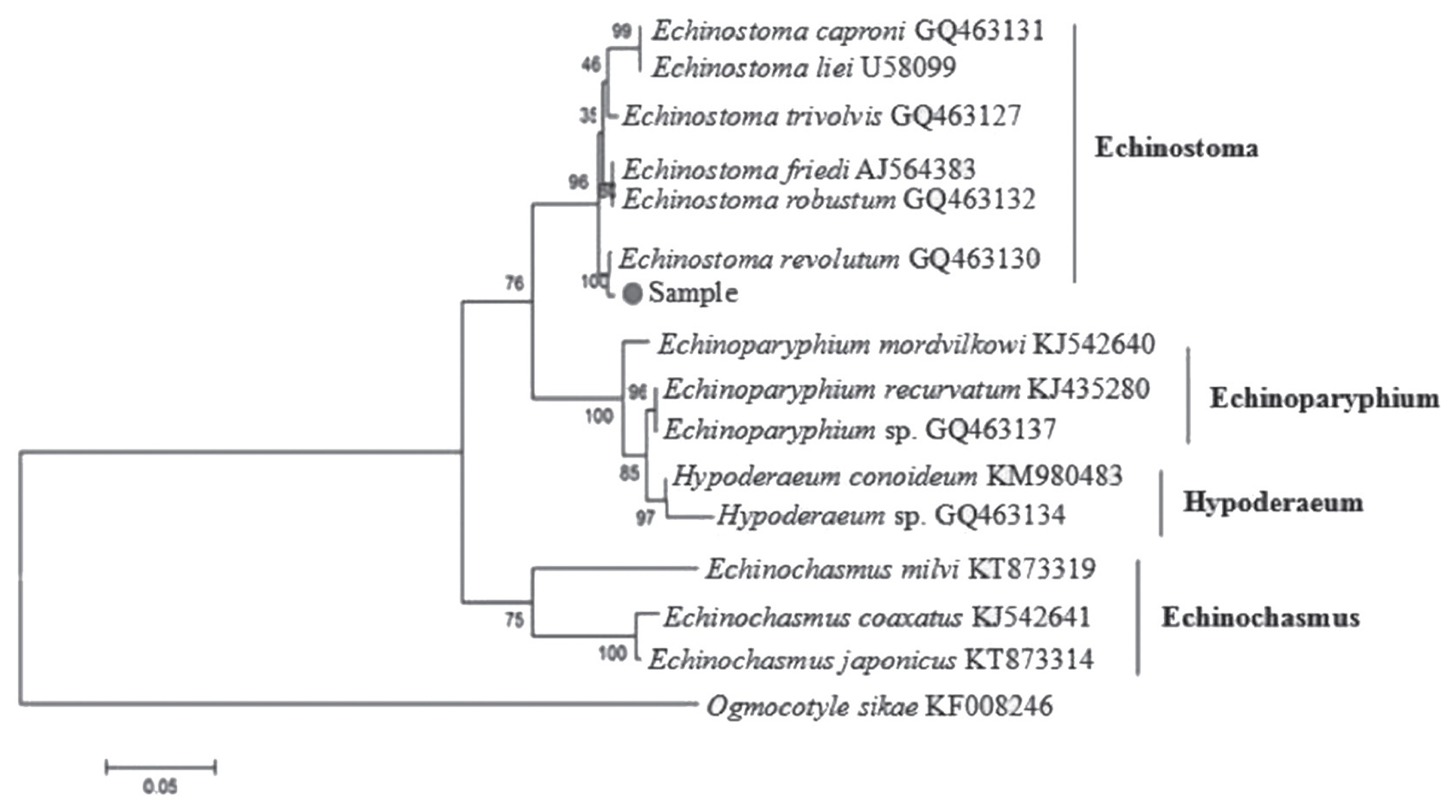
Fig. 2Arrangement of the mitochondrial genome of E. revolutum. All genes are predicted to be transcribed in clockwise direction. tRNA genes are designated by single-letter abbreviations for the corresponding amino acids.
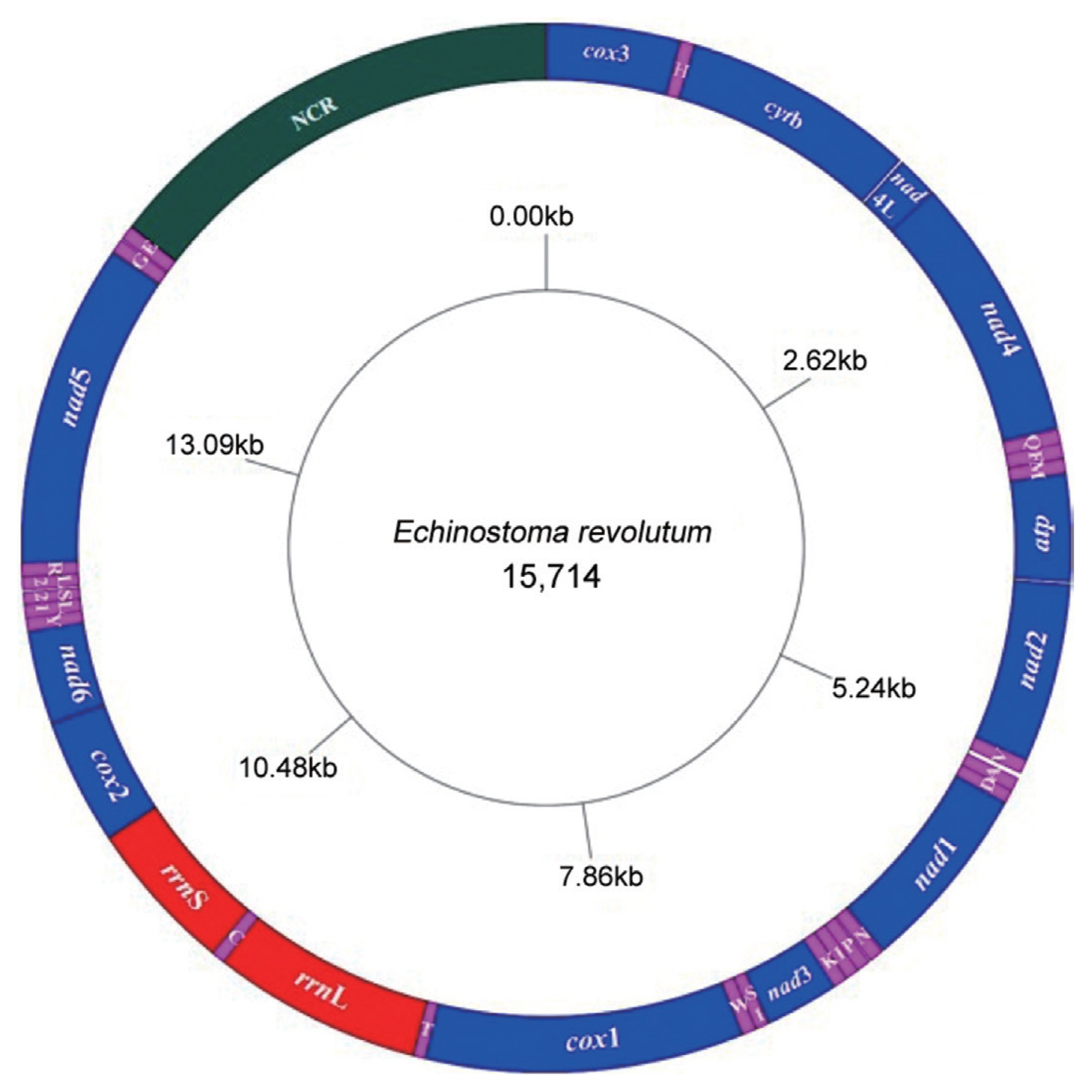
Table 1Organization of Echinostoma revolutum mitochondrial genome
Table 1
|
Gene/Region |
Position (Fragment size) |
Codon (Start/Stop) |
Anticodon |
Intergenic nucleotides |
|
cox3 |
1–645 (645) |
ATG/TAG |
|
3 |
|
tRNA-His |
649–715 (67) |
|
GUG |
2 |
|
cytb |
718–1,833 (1116) |
ATG/TAG |
|
2 |
|
nad4L |
1,836–2,108 (273) |
GTG/TAA |
|
−38 |
|
nad4 |
2,069–3,352 (1284) |
ATG/TAA |
|
7 |
|
tRNA-Gln |
3,360–3,426 (67) |
|
GUC |
0 |
|
tRNA-Phe |
3,427–3,493 (67) |
|
GAA |
7 |
|
tRNA-Met |
3,501–3,568 (68) |
|
UGG |
3 |
|
atp6 |
3,572–4,090 (519) |
ATG/TAG |
|
11 |
|
nad2 |
4,102–4,968 (867) |
GTG/TAG |
|
0 |
|
tRNA-Val |
4,969–5,033 (65) |
|
UAC |
29 |
|
tRNA-Ala |
5,063–5,131 (69) |
|
UGC |
1 |
|
tRNA-Asp |
5,133–5,195 (63) |
|
GUC |
1 |
|
nad1 |
5,197–6,114 (918) |
GTG/TAG |
|
7 |
|
tRNA-Asn |
6,122–6,191 (70) |
|
GUU |
8 |
|
tRNA-Pro |
6,200–6,267 (68) |
|
UGG |
0 |
|
tRNA-Ile |
6,268–6,332 (65) |
|
GAU |
3 |
|
tRNA-Lys |
6,336–6,403 (68) |
|
CUU |
0 |
|
nad3 |
6,404–6,760 (357) |
ATG/TAG |
|
3 |
|
tRNA-SerGCU
|
6,764–6,822 (59) |
|
GCU |
8 |
|
tRNA-Trp |
6,831–6,896 (66) |
|
UCA |
3 |
|
cox1 |
6,900–8,438 (1,539) |
GTG/TAG |
|
0 |
|
tRNA-Thr |
8,439–8,502 (64) |
|
GUU |
0 |
|
rrnL |
8,503–9,513 (1,011) |
|
|
0 |
|
tRNA-Cys |
9,514–9,578 (65) |
|
GCA |
0 |
|
rrnS |
9,579–10,321 (743) |
|
|
0 |
|
cox2 |
10,322–10,930 (609) |
ATG/TAA |
|
2 |
|
nad6 |
10,933–11,385 (453) |
GTG/TAG |
|
1 |
|
tRNA-Tyr |
11,387–11,450 (64) |
|
GUC |
0 |
|
tRNA-LeuAUG
|
11,451–11,514 (64) |
|
AUG |
2 |
|
tRNA-SerAGU
|
11,517–11,579 (63) |
|
AGU |
5 |
|
tRNA-LeuUAA
|
11,585–11,647 (63) |
|
|
2 |
|
tRNA-Arg |
11,650–11,712 (63) |
|
|
1 |
|
nad5 |
11,714–13,282 (1,569) |
|
GTG/TAA |
7 |
|
tRNA-Gly |
13,290–13,356 (67) |
|
|
6 |
|
tRNA-Glu |
13,363–13,431 (69) |
|
|
3 |
|
NCR |
13,435–15,714 (2,280) |
|
|
0 |
References
- 1. Chai JY. Echinostomes in humans. In: Fried B, Toledo R, editors. The Biology of Echinostomes: From the Molecule to the Community. New York, USA. Springer; 2009.
- 2. Chai JY. Intestinal flukes. In: Murrell KD, Fried B, editors. Food-Borne Parasitic Zoonoses: Fish and Plant-Borne Parasites, World Class Parasites. 11:New York, USA. Springer; 2007. p. 53-115.
- 3. Chai JY, Shin EH, Lee SH, Rim HJ. Foodborne intestinal flukes in Southeast Asia. Korean J Parasitol 2009;47:69-102.
- 4. Sohn WM, Chai JY, Yong TS, Eom KS, Yoon CH, Sinuon M, Socheat D, Lee SH.
Echinostoma revolutum, infection in children, Pursat Province, Cambodia. Emerg Inf Dis 2011;17:117-119.
- 5. Fried B, Graczyk TK. Recent advances in the biology of Echinostoma species in the “revolutum” group. Adv Parasitol 2004;58:139-145.
- 6. Kostadinova A, Gibson DI, Biserkov V, Chipev N. Re-validation of Echinostoma miyagawai Ishii, 1932 (Digenea: Echinostomatidae) on the basis of the experimental completion of its life-cycle. Syst Parasitol 2000;45:81-92.
- 7. Yang F, Zhang P, Shi XL, Li KX, Wang MW, Fu YQ, Yan XX, Hang JX, Li GQ. New record of Ascaridia nymphii (Secernentea: Ascaridiidae) from macaw parrot, Ara chloroptera, in China. Parasitol Int 2018;67:309-312.
- 8. Saijuntha W, Sithithaworn P, Duenngai K. Genetic variation and relationships of four species of medically important echinostomes (Trematoda: Echinostomatidae) in South-East Asia. Infect Genet Evol 2011;11:375-381.
- 9. Okimoto R, Macfarlane JL, Wolstenholme DR. Evidence for the frequent use of TTG as the translation initiation codon of mitochondrial protein genes in the nematodes, Ascaris suum and Caenorhabditis elegans
. Nucl Acids Res 1990;18:6-8.
- 10. Boore JL. Animal mitochondrial genomes. Nucl Acids Res 1999;27:1767-1780.
- 11. Jex AR, Hu M, Littlewood DT, Waeschenbach A, Gasser RB. Using 454 technology for long-PCR based sequencing of the complete mitochondrial genome from single Haemonchus contortus (Nematoda). BMC Genomics 2008;9:11-13.
- 12. Na L, Gao JF, Liu GH, Fu X, Su X, Yue DM, Gao Y, Zhang Y, Wang CR. The complete mitochondrial genome of Metorchis orientalis (Trematoda: Opisthorchiidae): comparison with other closely related species and phylogenetic implications. Infect Genet Evol 2016;39:45-50.
- 13. Ma J, He JJ, Liu GH, Blair D, Liu LZ, Liu Y, Zhu XQ. Mitochondrial genome of Ogmocotyle sikae and implications for phylogenetic studies of the Notocotylidae trematodes. Infect Genet Evol 2016;37:208-214.
- 14. Liu ZX, Zhang Y, Liu YT, Chang QC, Su X, Fu X, Yue DM, Gao Y, Wang CR. Complete mitochondrial genome of Echinostoma hortense (Digenea: Echinostomatidae). Korean J Parasitol 2016;54:173-179.
- 15. Shi XL, Fu YQ, Abdullahi AY, Wang MW, Yang F, Yu XG, Pan WD, Yan XX, Hang JX, Zhang P, Li GQ. The mitochondrial genome of Ancylostoma tubaeforme from cats in China. J Helminthol 2018;92:22-33.
- 16. Zhang P, Ran RK, Abdullahi AY, Shi XL, Huang Y, Sun YX, Liu YQ, Yan XX, Hang JX, Fu YQ, Wang MW, Chen W, Li GQ. The mitochondrial genome of Dipetalonema gracile from squirrel monkey in China. J Helminthol 2018;94:e1.
- 17. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 2003;52:696-704.
- 18. Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony, version 4.0b10. Sinauer; Sunderland, Massachusetts, USA. 2002.
- 19. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011;28:2731-2739.
- 20. Chen YY, Chen S, Kang J, Fang H, Dao H, Guo WZ, Lai CH, Lai MY, Fan JH, Fu LC. Evolving molecular epidemiological profile of human immunodeficiency virus 1 in the southwest border of China. PLoS One 2014;9:e107578.
- 21. Le TH, Blair D, Mcmanus DP. Complete DNA sequence and gene organization of the mitochondrial genome of the liverfluke, Fasciola hepatica L. (Platyhelminthes; Trematoda). Parasitology 2001;123:609-621.
- 22. Le TH, Blair D, Agatsuma T, Humair PF, Campbell NJ, Iwagami M, Littlewood DT, Peacock B, Johnston DA, Bartley J, Rollinson D, Herniou EA, Zarlenga DS, McManus DP. Phylogenies inferred from mitochondrial gene orders - A cautionary tale from the parasitic flatworms. Mol Biol Evol 2001;17:1123-1125.
- 23. Le TH, Nguyen NTB, Nguyen KT, Doan HTT, Dung DT, Blair D. A complete mitochondrial genome from Echinochasmus japonicus supports the elevation of Echinochasminae Odhner, 1910 to family rank (Trematoda: Platyhelminthes). Infect Genet Evol 2016;45:369-377.
- 24. Cai XQ, Liu GH, Song HQ, Wu CY, Zhou FC, Yan HK, Yuan ZG, Lin RQ, Zhu XQ. Sequences and gene organization of the mitochondrial genomes of the liver flukes Opisthorchis viverrini and Clonorchis sinensis (Trematoda). Parasitol Res 2012;110:235-243.
- 25. Jia WZ, Yan HB, Ni XW, Lou ZZ, Li HM, Cao P, Cai XP. Advances in the study of helminth mitochondrial genomes and their associated applications. Chinese Sci Bul 2012;57:54-67.
- 26. Jia WZ, Yan HB, Shi WG, Guo AJ, Zhan F. Research progress on complete mitochondrial genome sequences and their application for trematodes. Chinese J Vet Sci 2011;31:926-932.
- 27. Ojala D, Montoya J, Attardi G. TRNA punctuation model of RNA processing in human mitochondria. Nature 1981;290:470-474.