Abstract
Trichomonas vaginalis is a flagellated protozoan that causes trichomoniasis, a common nonviral sexually transmitted infection. T. vaginalis infection is asymptomatic in most infected men but can lead to chronic infection. The inflammatory response to chronic T. vaginalis infection may contribute to prostatic diseases, such as prostatitis and benign prostatic hyperplasia (BPH); however, studies on the relationship between T. vaginalis infection and prostate diseases are scarce. In this review, we discuss evidence from our studies on the involvement of T. vaginalis in the pathogenesis of prostate diseases, such as prostatitis and BPH. Studies of prostatitis have demonstrated that the attachment of T. vaginalis trophozoite to prostate epithelial cells (PECs) induces inflammatory cytokine production and inflammatory cell migration, leading to prostatitis. T. vaginalis also causes pathological changes, such as inflammatory cell infiltration, acinar changes, interstitial fibrosis, and mast cell infiltration, in prostate tissues of infected rats. Thus, T. vaginalis is considered an infectious agent that triggers prostatitis. Meanwhile, studies of prostatic hyperplasia revealed that mast cells activated by T. vaginalis-infected prostate cells secreted inflammatory mediators, such as β-hexosaminidase and tryptase, which promoted proliferation of prostate stromal cell (PSC). Moreover, interleukin-6 produced by proliferating PSCs induced the multiplication of BPH-1 epithelial cells as a result of stromal–epithelial interaction, suggesting that the proliferation of T. vaginalis-infected prostate cells can be induced through crosstalk with mast cells. These collective findings suggest that T. vaginalis contributes to the progression of prostatitis and prostatic hyperplasia by creating an inflammatory microenvironment involving PECs and PSCs.
-
Key words: Trichomonas vaginalis, prostatitis, prostatic hyperplasia, inflammation, proliferation
Introduction
Trichomonas vaginalis is a flagellated protozoan belonging to the subphylum Mastigophora. Its total length, including the oval body, flagella, and axostyle, averages approximately 26 μm (21–32 μm). The body of
T. vaginalis measures 9.5×6.8 μm, and the undulating membrane extends 3/4th of its body length [
1].
T. vaginalis infects the urogenital system, causing trichomoniasis, and is the most common sexually transmitted protozoan. According to the World Health Organization estimates, the incidence of
T. vaginalis infection is over 142 million cases [
2]. In men, trichomoniasis causes pain during urination and ejaculation. Moreover, it increases the frequency of urination and results in a thin, white discharge from the penis as well as soreness, swelling, and redness around the head of the penis or foreskin. However, most
T. vaginalis infections are asymptomatic, remain undiagnosed and untreated, and are thought to result in chronic persistent prostatic infection. In addition, trichomoniasis is a risk factor for human immunodeficiency virus transmission [
3].
The prostate is a male urogenital organ located at the base of the bladder surrounding the urethra. It consists of 3 glandular zones, including the central, peripheral, and transition zones, along with a nonglandular region of the anterior prostate composed of fibromuscular stroma, which provides structural support [
4]. The glandular tissue contributes to reproductive function by secreting zinc, citrate, and proteins, such as prostatic acid phosphatase, β-microsemiprotein, and prostatic-specific antigen, into the seminal fluid [
5,
6], whereas the fibromuscular tissue aids in the compartmentalization of fluids during urination and ejaculation.
Sfanos et al. [
7] have suggested the existence of barrier functions and antimicrobial defenses in the prostate; epithelial cells in the prostate express pathogen pattern recognition receptors and Toll-like receptor 4 (TLR4), which may serve as defenses against prostate infections [
8,
9]. In addition, the prostatic fluid contains several antimicrobial proteins, such as lactoferrin, defensins, immunoglobulins, and complement proteins, as well as high levels of free zinc and citrate [
10–
12]. The strong antimicrobial activities of prostatic secretions may be important in the protection of the male genitourinary tract from ascending infections and sperm transiting into the female genital tract.
While microaerophilic
T. vaginalis survive well in the vagina, which is essentially a hollow cavity, their survival becomes increasingly difficult once they reach the prostate, which contains antimicrobial proteins as well as high concentrations of zinc and citrate. Nevertheless, Gardner et al. [
13] identified Tvs in the prostatic urethra, glandular lumina, submucosa, and stroma of 5 prostate glands obtained at autopsy via immunoperoxidase staining. The foci of nonspecific acute and chronic inflammation as well as intraepithelial vacuolization have also been reported to be associated with Tv infection. Moreover, Mitteregger detected Tv DNA in 34% of the prostate tissues obtained from 86 patients with benign prostatic hyperplasia (BPH), indicating that Tv is chronically present in BPH tissue and may influence prostate growth [
14]. In addition, Iqbal et al. [
15] detected
T. vaginalis DNA and antigen in the prostate tissues of 42 (25%) and 37 (22%) patients with BPH, respectively.
Chronic infection with T. vaginalis may affect the development of prostatic hyperplasia as well as prostatitis. In this review, we discuss the hypothesis that T. vaginalis is involved in the initiation and/or progression of prostate diseases based on the results of both in vitro and in vivo experiments.
Prostatitis
Prostatitis is the most common urological diagnosis in men below 50 years of age, accounting for 8% of all office visits to urologists [
16]. The most frequently diagnosed form of prostatitis is chronic prostatitis/chronic pelvic pain syndrome (CP/CPPS). However, despite its high prevalence, prostatitis remains poorly understood, and the majority of diagnosed cases are of unknown etiology. Predisposing factors for CP/CPPS include heredity, infection, voiding abnormalities, hormone imbalance, intraprostatic reflux, immunological/allergic triggers, and psychological factors. Infection is responsible for 71–74% of chronic prostatitis cases, with 11–19% cases resulting from trichomoniasis infection [
17,
18]. In Korea, the prevalence of
T. vaginalis infection, as determined via polymerase chain reaction, is 4 and 21% in patients with chronic prostatitis/urethritis and prostatic hyperplasia, respectively [
19,
20]. Despite these high prevalence rates, experimental induction of prostatic infection by
T. vaginalis has not been attempted; however, it was determined whether exposure to
T. vaginalis could induce an inflammatory response in normal prostate epithelial cells (PECs).
Alderete et al. [
21,
22] reported that the adhesion of
T. vaginalis to mucosal cells is the first and essential step toward infection. Electron microscopy studies revealed that in vitro-grown
T. vaginalis cells with a typical globular shape rapidly transform into flat and amoeboid cells upon contact with vaginal epithelial cells (VECs), maximizing the area of adhesion [
23,
24]. In contrast, little is known about the adherence of
T. vaginalis to PECs.
T. vaginalis stained with a CellTrackerOrange fluorescent probe was incubated with PECs (cell line RWPE-1) and observed under a fluorescence microscope. When PECs were incubated with
T. vaginalis at ratios of 1:0.4 to 1:4, the adhesion of
T. vaginalis began after 30 min and increased continuously for 24 h. When the number of trichomonads increased, the rate of adhesion to PECs also concomitantly increased [
25]. At the RWPE-1:
T. vaginalis ratio of 1:0.4, cytotoxic damage to PECs was evident at 9 h, whereas at the ratio of 1:4, it was observed at 3 h. Epithelial–mesenchymal transition (EMT) was confirmed by decreased E-cadherin expression and increased vimentin expression at 24 h [
25,
26], which was similar to that observed in type 2 EMT associated with wound healing and tissue regeneration.
When
T. vaginalis attaches to PECs, it stimulates the production of IL-1β, IL-6, CCL2, and CXCL8 by activating ROS, ERK, and NF-κB signaling. Also, it has been reported that a PEC-conditioned medium containing these cytokines stimulates the migration of human neutrophils and monocytes. These data suggest that
T. vaginalis induces an inflammatory response in PECs [
27]. Because IL-1β and IL-6 play important roles in the progression of prostatic diseases, the signaling pathways involved in the production of these cytokines during
T. vaginalis infection of PECs were examined.
The NLRP3 inflammasome is involved in the production of IL-1β, a mediator of prostate inflammation associated with BPH, chronic prostatitis, and chronic pelvic pain syndrome [
28,
29]. The secretion of IL-1β is tightly regulated [
30]. First, pro-IL-1β is produced following the activation of pattern recognition receptors. This precursor is subsequently cleaved into a mature form by the proinflammatory cysteine protease caspase-1. Caspase-1 is activated in response to infection or tissue damage and is modulated by a macromolecular protein complex known as the inflammasome, which consists of a NOD-like receptor (NLR) family member, an adaptor protein, and an inactive caspase-1 precursor [
31]. The NLRP3 inflammasome has been implicated in antibacterial, viral, fungal, and parasitic immune responses [
32]. When PECs are infected with live
T. vaginalis, the expression of ASC, NLRP3, and caspase-1, all of which are components of the NLRP3 inflammasome, increases. Conversely, IL-1β production is decreased through siRNA-targeting of NLRP3 and caspase-1, confirming the involvement of the NLRP3 inflammasome in IL-1β production [
33].
IL-6 is a well-known biomarker of chronic prostatic inflammation and a mediator of chronic inflammation in prostate cancer [
34,
35]. Sutcliffe et al. [
36] suggested that IL-6 production in
T. vaginalis-exposed PECs promotes prostate carcinogenesis through the expression of proto-oncogenes, such as PIM1, c-MYC, and HMGA1; however, nothing is known regarding the signal pathways involved in IL-6 production in response to
T. vaginalis. Stimulation of PECs by
T. vaginalis resulted in the increased production of IL-6; increased expression of TLR2, TLR4, MAPKs, NF-κB, and JAK2/STAT3; and elevated levels of ROS. The inhibition of TLR2 or TLR4 reduced the production of IL-6 as well as the expression of other factors, and inhibitors of ROS, MAPKs, NF-κB, and JAK reduced IL-6 production. PECs stimulated with
T. vaginalis underwent EMT, which was suppressed by the inhibitors of JAK or NF-κB. That is,
T. vaginalis stimulates PECs to induce IL-6 production by activating TLR2/4, ROS, MAPKs, NF-κB, AP-1, JAK2/STAT3, and EMT via the NF-κB and JAK/STAT3 signaling pathways. Therefore,
T. vaginalis may alter the prostate tumor microenvironment by inducing IL-6 production and EMT [
26].
Prostatitis is caused by inflammatory responses that occurs not only in PECs but also in stromal cells.
T. vaginalis has been detected in the submucosal layer and stroma of the prostate through immunoperoxidase staining [
13]. The stromal compartment of the prostate primarily consists of smooth muscle cells, fibroblasts, macrophages, endothelial cells, and other immune cells [
37]. The WPMY-1 cell line, isolated from normal human prostate stroma, is a myofibroblast line immortalized with SV40 large T antigen. Myofibroblasts play an important role in wound contraction in fibrotic diseases as an intermediate cell type between fibroblast and smooth muscle cell. Moreover, myofibroblasts are the dominant cell type associated with prostatic hypertrophy and contribute to prostate cancer progression and metastasis [
38].
Previous studies showed that when RWPE-1 cells were incubated with
T. vaginalis, cell viability at 24 h decreased concomitantly with an increase in the number of trichomonads, confirming that
T. vaginalis is cytotoxic to RWPE-1 [
25,
26]. Sfanos et al. [
7] suggested that epithelial cell death and consequent barrier function disruption lead to infiltration of stromal compartments by commensal or pathogenic organisms. It has been hypothesized that
T. vaginalis penetrates the epithelium, enters the stroma, and induces an inflammatory response. Therefore, it was determined whether
T. vaginalis induces an inflammatory response in stromal cells. Incubation of WPMY-1 with live
T. vaginalis increased the expression of the inflammatory chemokines CXCL8 and CCL2. In addition, TLR4, ROS, MAPK, and NF-κB expression increased, whereas the inhibitors of TLR4, ROS, MAPKs, and NF-κB decreased CXCL8 and CCL2 production. Conditioned medium derived from WPMY-1 cells incubated with
T. vaginalis stimulated the migration of human neutrophils and monocytes, suggesting that
T. vaginalis increases CXCL8 and CCL2 production by human prostate stromal cells by activating TLR4, ROS, MAPKs and NF-κB. This, in turn, attracts neutrophils and monocytes and results in an inflammatory response [
39].
The in vitro inflammatory responses induced by
T. vaginalis in PECs and stromal cells prompted us to determine whether
T. vaginalis causes prostatitis in an animal model. To mimic the route of
T. vaginalis infection in humans,
T. vaginalis was injected through the urethras of Wistar rats.
T. vaginalis trophozoites were observed in the prostate acini of the injected rats via immunohistochemistry. The prostate tissues exhibited elevated pathological scores, and 83% (5/6 samples) and 100% (6/6 samples) of the ventral and dorsolateral lobes (
n=6), respectively, were inflamed. Pathological changes, such as inflammatory cell infiltration, acinar changes, and interstitial fibrosis, were observed. The inflammation was more severe in the dorsolateral lobes than in the ventral lobes. Moreover, infiltration and degranulation of mast cells were observed at higher rates in the prostate sections of the
T. vaginalis-infected rats, and the prostate tissues of the injected rats had increased CCL2 levels. These findings provided the first evidence that
T. vaginalis infection in rats causes prostatitis [
40].
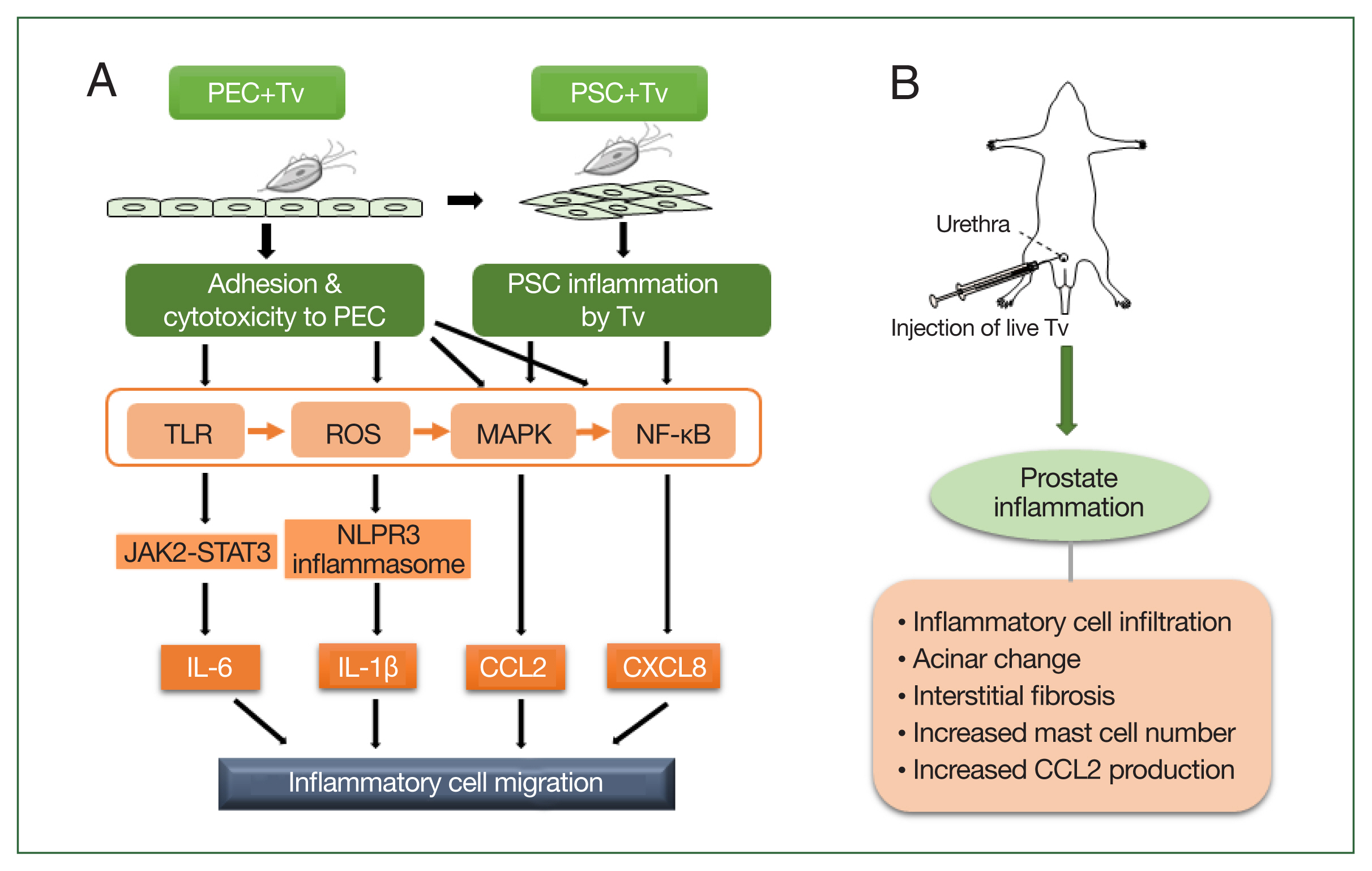
Taken together, the above results indicate that the mechanism by which
T. vaginalis causes prostatitis may occur as follows:
T. vaginalis causes prostatitis by attaching to prostate cells and inducing proinflammatory cytokine production, inflammatory cell migration, and EMT. In animal experiments, injection of
T. vaginalis into rats via the urethra caused prostatitis as evidenced by various pathologic changes, including inflammatory cell infiltration, acinar changes, interstitial fibrosis, mast cell infiltration, and increased CCL2 production. These in vitro and in vivo experimental evidence demonstrate that
T. vaginalis is an infectious agent causing prostatitis (
Fig. 1).
Prostate Hyperplasia
Benign prostate hyperplasia (BPH)
BPH results in the benign enlargement of the prostate gland due to the unregulated hyperplastic growth of the epithelial and fibromuscular tissues of the transition zone and periurethral area [
41]. BPH is an immune-mediated inflammatory disease, with persistent prostatic inflammation as a key factor in its development and progression [
42,
43]. An inflammatory reaction may be triggered by several factors, including bacterial infection (e.g.,
Escherichia coli), viruses (e.g., human papilloma virus, herpes simplex virus type 2, and cytomegalovirus), sexually transmitted organisms (e.g., gonorrhea and chlamydia), hormones, dietary factors, autoimmune response, and urinary reflux into the prostate collection ducts [
42,
44–
46]. The inflammatory response induced by these factors may cause T-lymphocyte infiltration, cytokine production, growth factor expression, local hypoxia with angiogenesis, tissue damage with abnormal wound healing and stromal and epithelial cell proliferation, and eventually BPH [
42,
45].
Several sexually transmitted micro organisms, such as
Neisseria gonorrhoeae, Chlamydia trachomatis, and
T. vaginalis, are known to cause chronic inflammation of the prostate tissue [
47]. However, in a study investigating the association between BPH (or lower urinary tract symptom-related outcomes) and antibodies against various sexually transmitted infections (e.g.,
C. trachomatis, T. vaginalis, human papilloma virus, herpes simplex virus, cytomegalovirus infection, gonorrhea, and syphilis), only
T. vaginalis infection was found to be linked to BPH [
48]. Moreover,
T. vaginalis DNA was detected in the prostate tissue and urine of patients with BPH, suggesting that it acts as an initial stimulus for an inflammatory response [
14,
15,
19]. In addition, Twu et al. [
48] reported that
T. vaginalis macrophage migration inhibitory factor induced an inflammatory response, prostate cell growth, and invasiveness in BPH-1 and prostate cancer cell lines.
To date, there have been few studies regarding prostate growth induced by the inflammatory response of prostate cells infected with live
T. vaginalis. Claus et al. [
50] reported that the proliferation rates of prostate epithelium and stroma increased by approximately 9-fold and 37-fold, respectively, in BPH compared with the normal prostate and that prostatic stromal hyperproliferation was an important feature of BPH pathogenesis. We focused on stromal hyperplasia in the experimental studies of prostatic hyperplasia.
We hypothesized that the crosstalk between prostatic cells inflamed by
T. vaginalis infection and migrating inflammatory cells was involved in the proliferation of prostatic cells. During inflammatory reaction in prostate tissue, T lymphocytes are the most common infiltrating cells. B lymphocytes and macrophages are also known to infiltrate [
42,
44]. However, clinical studies have suggested that mast cells play an important role in the development and persistence of inflammation in BPH associated with the lower urinary tract [
51–
53]. In our prostate proliferation studies, we focused on mast cells based on reports that they are involved in the inflammatory response to
T. vaginalis infection. Mast cells were preferentially detected in cervical smears from women with trichomoniasis compared with smears from patients with vaginitis caused by other pathogens [
54].
T. vaginalis (live trophozoites, excretory-secretory products [ESP]) was confirmed to induce an inflammatory response by migrating and activating mast cells, which resulted in the production of histamine and TNF-α [
55]. When VECs were cocultured with
T. vaginalis, VEC-produced inflammatory mediators activated and attracted mast cells and stimulated them to induce neutrophil migration [
56]. In addition, the number of mast cells and activated degranulated mast cells increased during prostatitis in
T. vaginalis-infected rats [
40].
Mast cells are important cellular sensors and regulators of inflammation, fibrosis, and smooth muscle cell contraction [
53,
57–
59]. Activated mast cells release various factors, such as chymase, tryptases, and proteases, which can interact with the local tissue environment and lead to tissue fibrosis, repair, and remodeling [
53]. Conversely, little is known regarding the role of mast cells in prostate proliferation resulting from
T. vaginalis infection.
T. vaginalis-infected BPH epithelial cells have been reported to produce cytokines, such as CXCL8, CCL2, IL-1β, and IL-6, through the ROS, MAPK, and NF-κB signaling pathways. Inflammatory mediators, including these cytokines, stimulate the migration of monocytes and mast cells. When mast cells are incubated with trichomonad-conditioned medium, the activated mast cells produce β-hexosaminidase and CXCL8 and induce PSC proliferation. Proliferation was decreased by inhibiting CXCR1 (receptor for CXCL8) and CCR2 (receptor for CCL2), indicating that CXCL8 and CCL2 stimulate PSC proliferation by binding to CXCR1 and CCR2, respectively. This study indicated that
T. vaginalis-infected BPH-1 cells activate mast cells, which release inflammatory mediators that induce PSC proliferation [
60,
61].
Induction of PSC proliferation by tryptase derived from activated mast cells
Tryptase is the most abundant serine proteinase found in the secretory granules of mast cells and has been reported to induce human lung fibroblast migration, myoblast proliferation, and cardiac fibroblast activation via protease-activated receptor 2 (PAR2) [
62–
64]. PAR2 is a G-protein-coupled receptor activated through proteolytic cleavage by serine proteases, including tryptase. PAR2 may be involved in tissue remodeling by inducing fibroblast migration, differentiation, and extracellular matrix production [
65]. Roman et al. [
66] suggested that PAR2 activation in the prostate contributes to the development of lower urinary tract dysfunction through proinflammatory as well as profibrotic pathways. Furthermore, activation of the tryptase–PAR2 axis is important for the onset of fibrosis.
Studies have examined whether tryptase released from mast cells activated by
T. vaginalis-infected prostate stromal cells (PSC) promotes the proliferation of PSCs via protease-activated receptor 2 (PAR2). Inflammatory mediators derived from
T. vaginalis-infected PSCs induced mast cell migration, tryptase production, and PSC proliferation. Signaling molecules involved in the tryptase–PAR2 pathway (e.g., PAR2, p-ERK, COX-2, 15d-PGJ2, and PPARγ) were increased, and the inhibition of tryptase and signaling molecules inhibited proliferation. These results indicate that the interaction between
T. vaginalis-infected PSCs and mast cells induces the proliferation of PSCs via the tryptase–PAR2 pathway [
67].
According to the American Urological Association, BPH is defined as a histologic diagnosis and refers to the proliferation of smooth muscle and epithelial cells within the prostatic transition zone [
68]. Although there are reports regarding the proliferation of prostate stromal cells, only limited information is available on the proliferation of PECs.
Stromal–epithelial interactions play an important regulatory role in the development of the prostate and in the maintenance of the adult prostate in health and disease [
69]. Siejka et al. [
70] reported that bidirectional stromal–epithelial interactions occur in the prostate gland. When the supernatants of prostate stromal cells (or BPH epithelial cells) were added to BPH epithelial cells (or prostate stromal cells), proliferation was increased. In BPH tissues, the ratio of stromal to epithelial cells reached a value of 5.
As discussed in Proliferation of stromal cells is induced by crosstalk between
T. vaginalis-infected BPH epithelial cells and mast cells, the proliferation of prostate stromal cells is induced by BPH epithelial cells in response to
T. vaginalis infection through crosstalk with mast cells. Subsequently, the effect of IL-6 released by proliferating stromal cells on the induction of BPH epithelial cells was determined. When culture supernatants of proliferating prostate stromal cells were added to BPH epithelial cells, the latter multiplied and cyclin D1, FGF2, and Bcl-2 expression increased. Blocking the IL-6 signaling pathway with anti-IL-6R antibody or a JAK1/2 inhibitor inhibited the proliferation of BPH epithelial cells and reduced the expression of IL-6, IL-6R, and STAT3. EMT was also detected in proliferating BPH epithelial cells, indicating that IL-6 released from proliferating prostate stromal cells induced by BPH epithelial cells infected with
T. vaginalis promoted BPH epithelial cell multiplication [
71]. This indicates that the inflammatory microenvironment of prostate stromal cells resulting from
T. vaginalis infection promotes the proliferation of PECs through stromal–epithelial interaction.
EMT is a complex process that involves the transformation of epithelial cells into stromal cells. It is integral to development, wound healing, and stem cell behavior, and it contributes to fibrosis and cancer progression [
72,
73]. Alonso-Magdalena et al. [
74] analyzed prostate samples from 16 patients with BPH. BPH is not a disease of the prostate stroma but is the accumulation of mesenchymal-like cells derived from the prostate epithelium, suggesting that EMT plays an important role in the development of BPH. Shi et al. [
75] and Hu et al. [
76] reported that estradiol and TGF-B1/Smad induce EMT in BPH epithelial cells. Thus, EMT in BPH epithelial cells proliferating in response to IL-6 produced by
T. vaginalis-infected stromal cells is involved in the pathogenesis of BPH.
BPH may be linked to obesity as abdominal obesity and serum leptin levels were found to be associated with prostate growth in a retrospective cohort study in Korea [
77]. Male obesity has also been linked to increased severity of lower urinary tract symptoms (LUTS) in men affected by BPH [
78]. Adipocytes, also known as lipocytes or fat cells, are the main components of adipose tissue that specialize in storing energy as fat. Adipokines produced by adipose tissue affect metabolic processes and mediate inflammation, cell proliferation, and angiogenesis [
79]. Leptin is an adipokine originally identified as a key molecule regulating food intake and body weight. It interacts with other hormones and energy regulators to mediate the effects of insulin, glucagon, insulin-like growth factor, growth hormone, glucocorticoids, cytokines, and metabolites [
80]. Leptin also stimulates the growth of prostate, breast, lung, ovarian, and pancreatic cancer cells [
81]. Prostate cells proliferate when leptin is administered together with estrogen; however, reports of prostate cell proliferation in response to leptin alone are rare [
82].
A study was performed to determine whether
T. vaginalis-infected BPH epithelial cells (BPH-1 cells) induced the proliferation of prostate cells through a leptin signaling pathway. BPH-1 cells incubated with live
T. vaginalis released proinflammatory cytokines, and conditioned medium from these cells stimulated adipocyte migration. When PSCs and BPH-1 cells were incubated with adipocyte-conditioned medium containing leptin (ATCM), their growth rates increased. In addition, the expression of the leptin receptor (known as OBR) and downstream signaling molecules, such as JAK2/STAT3, Notch1/Jagged1, and survivin, increased. Moreover, OBR blocking reduced proliferation and the expression of leptin signaling molecules in response to ATCM. These findings indicate that inflamed BPH-1 cells infected with
T. vaginalis promote prostate cell proliferation through leptin–OBR signaling. Therefore, it is likely that
T. vaginalis contributes to prostate enlargement in BPH via adipocyte leptin release as a result of prostate inflammation [
83].
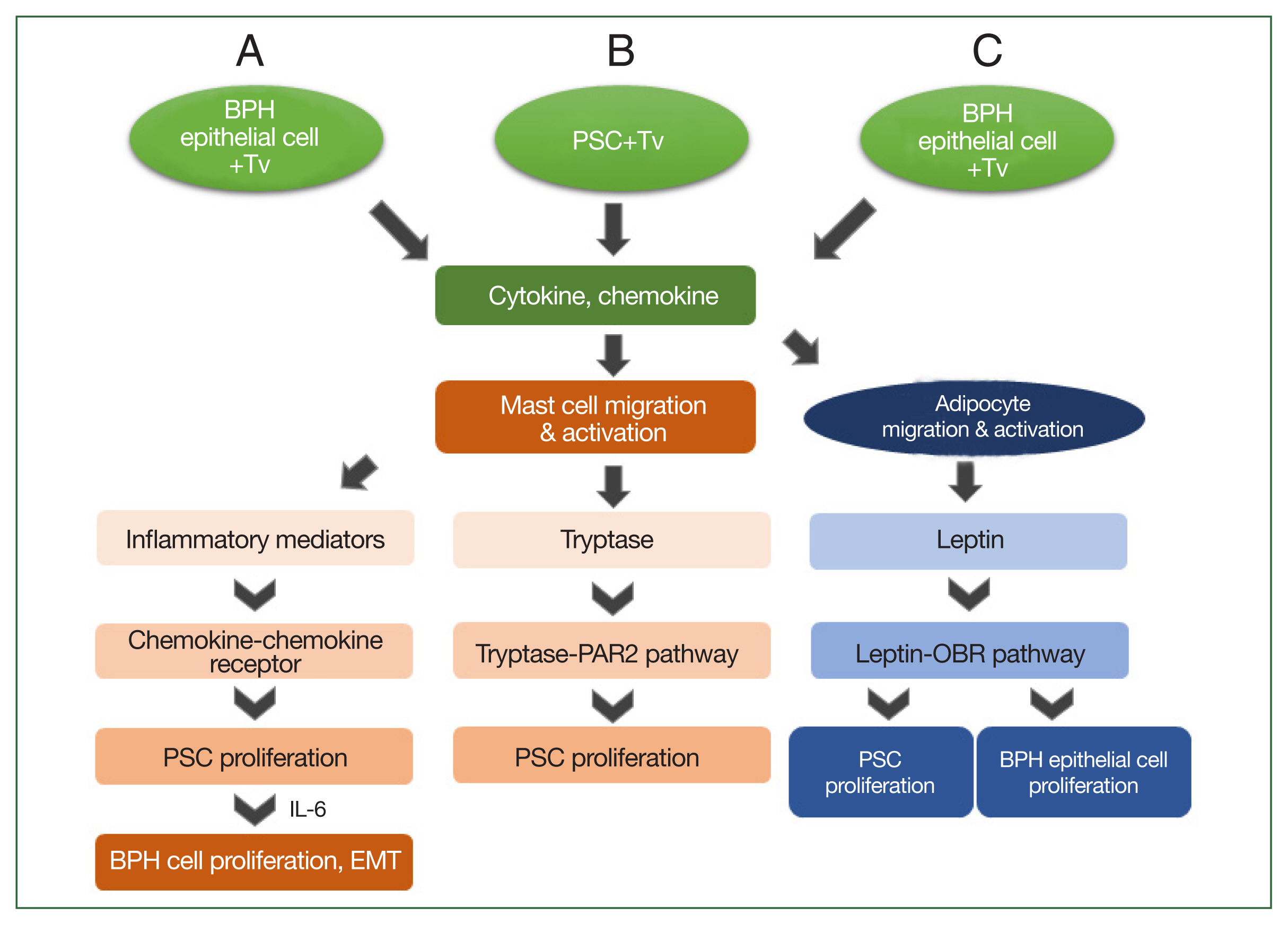
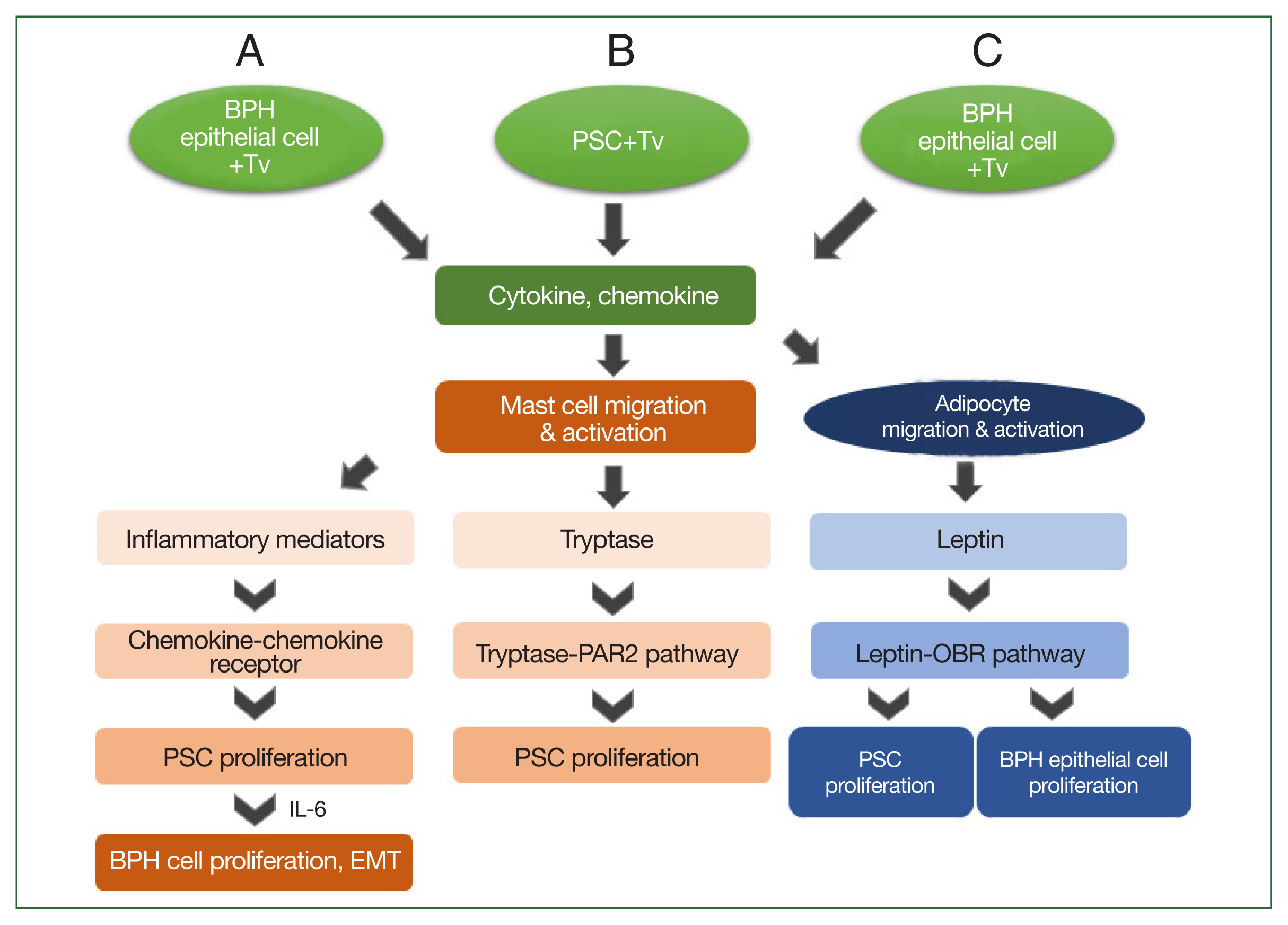
Based on the above results, the mechanism by which
T. vaginalis induces prostate cell proliferation may be as follows: when
T. vaginalis interacts with prostate cells, it triggers an inflammatory response, and migratory mast cells induce PSC proliferation by secreting inflammatory mediators or producing tryptase. Subsequently, the proliferating PSCs produce IL-6, which stimulates BPH-1 epithelial cell proliferation. These findings indicate that bidirectional stromal–epithelial interactions are responsible for PEC proliferation. In addition, when BPH-1 cells are infected with
T. vaginalis, adipocytes migrate and produce leptin, which results in the proliferation of prostate cells through the leptin–OBR signaling pathway (
Fig. 2). Taken together, it appears that
T. vaginalis causes proliferation of the prostate via the infiltration of inflammatory cells, including mast cells and adipocytes.
Conclusion
When T. vaginalis interacts with prostate cells, such as PECs, prostate stromal cells, or BPH epithelial cells, inflammatory mediators such as cytokines are produced to create an inflammatory microenvironment. In addition, the inflammatory mediators released from activated inflammatory cells, including migratory mast cells and adipocytes, amplify the inflammatory response via cell–cell interactions, leading to prostatitis or BPH. Although this review highlights some pathogen-induced prostate inflammatory responses, more studies in this field are needed.
Notes
-
Author contributions
Conceptualization: Ryu JS
Data curation: Kim JH
Investigation: Han IH, Kim JH, Ryu JS
Methodology: Han IH, Kim JH
Supervision: Ryu JS
Writing – original draft: Han IH, Ryu JS
-
The authors declare no conflict of interest related to this study.
Acknowledgments
We thank Min-Young Seo, Su-Jeong Lim, Na-Young Gu, Sang-Su Kim, Hyo-Yeoung Chung, Dr. Kyu-Shik Kim, Dr. Ik-Hwan Han, Dr. Jung-Hyun Kim, and the late Dr. Ki-Seok Jang for participating in the experiments. Also we would like to thank Professor Soon-Jung Park for revising this review.
Fig. 1Schematic diagram of the induction of prostatitis by Trichomonas vaginalis (Tv). (A) Tv attaches to prostate epithelial cells (PECs) and causes an inflammatory response, resulting in the increased production of cytokines, such as IL-1β, IL-6, CXCL8, and CCL2. In addition, Tv causes cytotoxic damage to epithelial cells, thereby infecting prostate stromal cells (PSCs) and triggering an inflammatory response that induces the migration of inflammatory cells. TLR, ROS, MAPK, and NF-kB are generally involved in cytokine production, and in particular, the NLRP3 inflammasome and JAK2-STAT3 are implicated in IL-1β and IL-6 production, respectively. (B) Injection of live Tv through the rat urethra induces prostatitis.
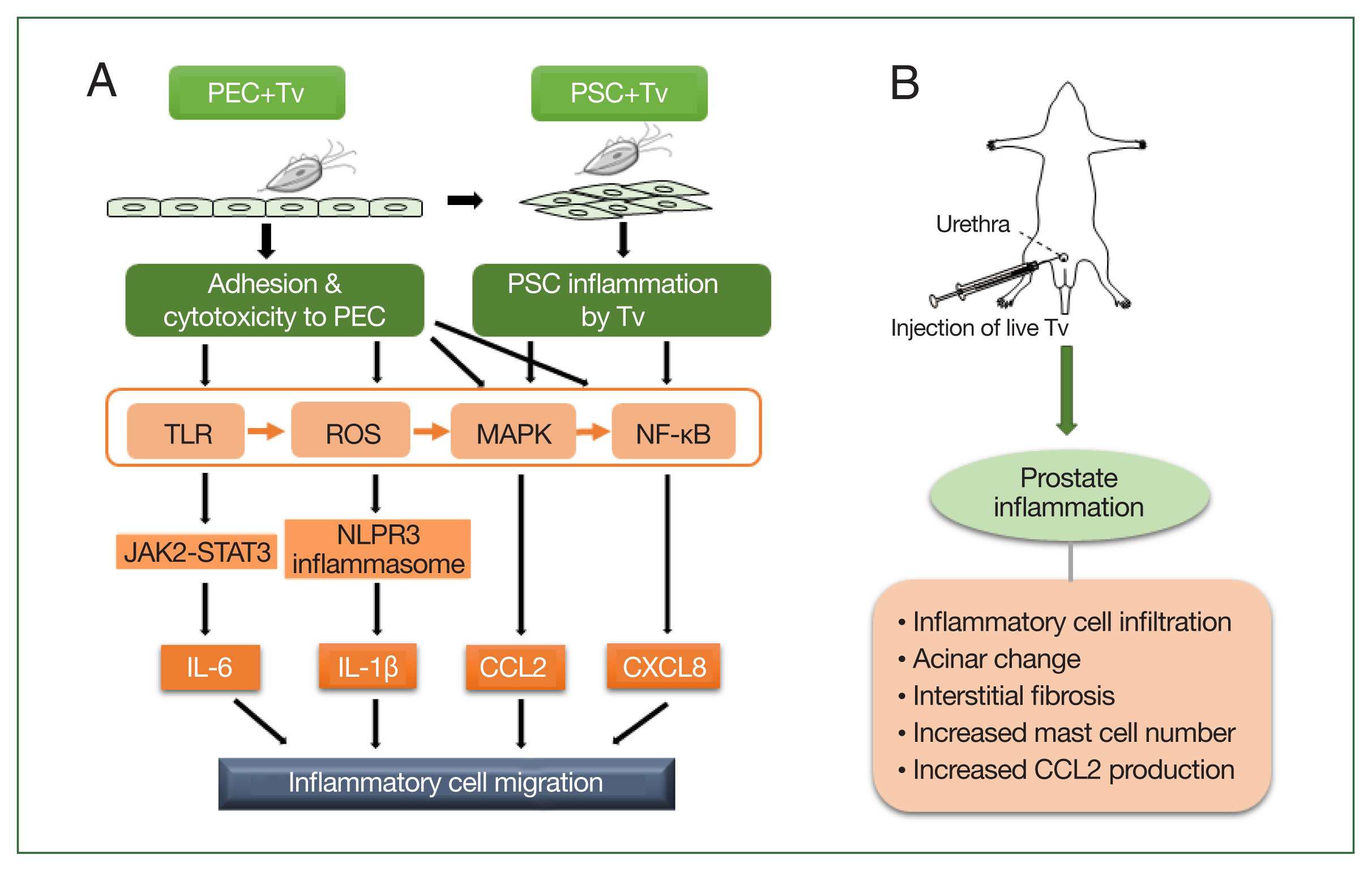
Fig. 2Schematic diagram of the induction of benign prostatic hyperplasia (BPH) by Trichomonas vaginalis (Tv). When BPH-1 epithelial cells and prostate stromal cells (PSCs) interact with Tv, they produce cytokines and chemokines that activate mast cells or adipocytes and stimulate their migration. (A) For BPH-1 cells, chemokines from activated mast cells bind to a chemokine receptor on PSCs to induce their proliferation. Subsequently, the proliferating PSCs produce IL-6, which results in BPH-1 epithelial cell proliferation. (B) Tryptase released from mast cells activated by Tv-infected PSCs promotes the proliferation of PSCs through the tryptase–protease-activated receptor 2 (PAR2) signaling pathway. (C) Cytokines and chemokines released by Tv-infected BPH-1 cells trigger adipocyte migration and activation. Leptin produced by activated adipocytes induces prostate cell proliferation through the leptin–OBR signaling pathway.
References
- 1. Cheon SH, Kim SR, Song HO, Ahn MH, Ryu JS. The dimension of Trichomonas vaginalis as measured by scanning electron microscopy. Korean J Parasitol 2013;51(2):243-246. http://doi.org/10.3347/kjp.2013.51.2.243
- 2. World Health Organization. Global health sectors strategy on sexually transmitted infections 2016–2021. World Health Organization; Geneva, Switzerland. 2016.
- 3. McClelland RS, Sangare L, Hassan WM, Lavreys L, Mandaliya K, et al. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J Infect Dis 2007;195(5):698-702. http://doi.org/10.1086/511278
- 4. McNeal JE. Anatomy of the prostate and morphogenesis of BPH. Prog Clin Biol Res 1984;145:27-53.
- 5. Leach DA, Need EF, Toivanen R, Trotta AP, Palethorpe HM, et al. Stromal androgen receptor regulates the composition of the microenvironment to influence prostate cancer outcome. Oncotarget 2015;6(18):16135-16150. http://doi.org/10.18632/oncotarget.3873
- 6. Lilja H, Abrahamsson PA. Three predominant proteins secreted by the human prostate gland. Prostate 1988;12(1):29-38. http://doi.org/10.1002/pros.2990120105
- 7. Sfanos KS, Yegnasubramanian S, Nelson WG, De Marzo AM. The inflammatory microenvironment and microbiome in prostate cancer development. Nat Rev Urol 2018;15(1):11-24. http://doi.org/10.1038/nrurol.2017.167
- 8. Gatti G, Quintar AA, Andreani V, Nicola JP, Maldonado CA, et al. Expression of Toll-like receptor 4 in the prostate gland and its association with the severity of prostate cancer. Prostate 2009;69(13):1387-1397. http://doi.org/10.1002/pros.20984
- 9. Gambara G, De Cesaris P, De Nunzio C, Ziparo E, Tubaro A, et al. Toll-like receptors in prostate infection and cancer between bench and bedside. J Cell Mol Med 2013;17(6):713-722. http://doi.org/10.1111/jcmm.12055
- 10. Manning ML, Williams SA, Jelinek CA, Kostova MB, Denmeade SR. Proteolysis of complement factors iC3b and C5 by the serine protease prostate-specific antigen in prostatic fluid and seminal plasma. J Immunol 2013;190(6):2567-2574. http://doi.org/10.4049/jimmunol.1200856
- 11. Fowler JE Jr, Mariano M. Longitudinal studies of prostatic fluid immunoglobulin in men with bacterial prostatitis. J Urol 1984;131(2):363-369. http://doi.org/10.1016/s0022-5347(17)50387-0
- 12. Isaacs JT. Prostatic structure and function in relation to the etiology of prostatic cancer. Prostate 1983;4(4):351-366. http://doi.org/10.1002/pros.2990040405
- 13. Gardner WA Jr, Culberson DE, Bennett BD. Trichomonas vaginalis in the prostate gland. Arch Pathol Lab Med 1986;110(5):430-432.
- 14. Mitteregger D, Aberle SW, Makristathis A, Walochnik J, Brozek W, et al. High detection rate of Trichomonas vaginalis in benign hyperplastic prostatic tissue. Med Microbiol Immunol 2012;201(1):113-116. http://doi.org/10.1007/s00430-011-0205-2
- 15. Iqbal J, Al-Rashed J, Kehinde EO. Detection of Trichomonas vaginalis in prostate tissue and serostatus in patients with asymptomatic benign prostatic hyperplasia. BMC Infect Dis 2016;16(1):506. http://doi.org/10.1186/s12879-016-1843-1
- 16. Thumbikat P, Shahrara S, Sobkoviak R, Done J, Pope RM, et al. Prostate secretions from men with chronic pelvic pain syndrome inhibit proinflammatory mediators. J Urol 2010;184(4):1536-1542. http://doi.org/10.1016/j.juro.2010.05.086
- 17. Skerk V, Schönwald S, Krhen I, Markovinović L, Beus A, et al. Aetiology of chronic prostatitis. Int J Antimicrob Agents 2002;19(6):471-474. https://doi.org/10.1016/S0924-8579(02)00087-0
- 18. Skerk V, Krhen I, Schonwald S, Cajic V, Markovinovic L, et al. The role of unusual pathogens in prostatitis syndrome. Int J Antimicrob Agents 2004;24(suppl 1):53-56. https://doi.org/10.1016/j.ijantimicag.2004.02.010
- 19. Lee JJ, Moon HS, Lee TY, Hwang HS, Ahn MH, et al. PCR for diagnosis of male Trichomonas vaginalis infection with chronic prostatitis and urethritis. Korean J Parasitol 2012;50(2):157-159. https://doi.org/10.3347/kjp.2012.50.2.157
- 20. Seo JH, Yang HW, Joo SY, Song SM, Lee YR, et al. Prevalence of Trichomonas vaginalis by PCR in men attending a primary care urology clinic in South Korea. Korean J Parasitol 2014;52(5):551-555. https://doi.org/10.3347/kjp.2014.52.5.551
- 21. Alderete JF, Pearlman E. Pathogenic Trichomonas vaginalis cytotoxicity to cell culture monolayers. Br J Vener Dis 1984;60 (2):99-105. https://doi.org/10.1136/sti.60.2.99
- 22. Alderete JF, Garza GE. Specific nature of Trichomonas vaginalis parasitism of host cell surfaces. Infect Immun 1985;50(3):701-708. https://doi.org/10.1128/iai.50.3.701-708.1985
- 23. Arroyo R, Gonzalez-Robles A, Martinez-Palomo A, Alderete JF. Signalling of Trichomonas vaginalis for amoeboid transformation and adhesion synthesis follows cytoadherence. Mol Microbiol 1993;7(2):299-309. https://doi.org/10.1111/j.1365-2958.1993.tb01121.x
- 24. Kim SR, Ryu JS. Scanning electron microscopic observation of Trichomonas vaginalis contacted with human vaginal epithelial cells. Korean J Electron Microscopy 2001;31(3):235-244. (in Korean).
- 25. Kim JH, Han IH, Kim SS, Park SJ, Min DY, et al. Interaction between Trichomonas vaginalis and the Prostate Epithelium. Korean J Parasitol 2017;55(2):213-218. https://doi.org/10.3347/kjp.2017.55.2.213
- 26. Han IH, Kim JH, Kim SS, Ahn MH, Ryu JS. Signalling pathways associated with IL-6 production and epithelial-mesenchymal transition induction in prostate epithelial cells stimulated with Trichomonas vaginalis. Parasite Immunol 2016;38(11):678-687. https://doi.org/10.1111/pim.12357
- 27. Seo MY, Im SJ, Gu NY, Kim JH, Chung YH, et al. Inflammatory Response of Prostate Epithelial Cells to Stimulation by Trichomonas vaginalis. Prostate 2014;74:441-449. https://doi.org/10.1002/pros.22766
- 28. Kashyap M, Pore S, Wang Z, Gingrich J, Yoshimura N, et al. Inflammasomes are important mediators of prostatic inflammation associated with BPH. J Inflamm (Lond) 2015;12:37. https://doi.org/10.1186/s12950-015-0082-3
- 29. Chen CS, Chang PJ, Lin WY, Huang YC, Ho DR. Evidences of the inflammasome pathway in chronic prostatitis and chronic pelvic pain syndrome in an animal model. Prostate 2013;73 (4):391-397. https://doi.org/10.1002/pros.22580
- 30. Dinarello CA. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr 2006;83(2):447S-455S. https://doi.org/10.1002/pros.22580
- 31. Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004;117(5):561-574. https://doi.org/10.1016/j.cell.2004.05.004
- 32. Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the antimicrobial response by NLR proteins. Immunity 2011;34(5):665-679. https://doi.org/10.1016/j.immuni.2011.05.007
- 33. Gu NY, Kim JH, Han IH, Im SJ, Seo MY, et al. Trichomonas vaginalis induces IL-1 beta production in a human prostate epithelial cell line by activating the NLRP3 inflammasome via reactive oxygen species and potassium ion efflux. Prostate 2016;76(10):885-896. https://doi.org/10.1002/pros.23178
- 34. Nguyen DP, Li JY, Tewari AK. Inflammation and prostate cancer: the role of interleukin 6 (IL-6). BJU Int 2014;113(6):986-992. https://doi.org/10.1111/bju.12452
- 35. Bardan R, Dumache R, Dema A, Cumpanas A, Bucuras V. The role of prostatic inflammation biomarkers in the diagnosis of prostate diseases. Clin Biochem 2014;47(10–11):909-915. https://doi.org/10.1016/j.clinbiochem.2014.02.008
- 36. Sutcliffe S, Neace C, Magnuson NS, Reeves R, Alderete JF. Trichomonosis, a common curable STI, and prostate carcinogenesis-a proposed molecular mechanism. PLoS Pathog 2012;8(8):e1002801. https://doi.org/10.1371/journal.ppat.1002801
- 37. Thomson AA, Cunha GR, Marker PC. Prostate development and pathogenesis. Differentiation 2008;76(6):559-564. https://doi.org/10.1111/j.1432-0436.2008.00303.x
- 38. Tuxhorn JA, Ayala GE, Rowley DR. Reactive stroma in prostate cancer progression. J Urol 2001;166(6):2472-2483.
- 39. Im SJ, Han IH, Kim JH, Gu NY, Seo MY, et al. Inflammatory response of a prostate stromal cell line induced by Trichomonas vaginalis. Parasite Immunol 2016;38(4):218-227. https://doi.org/10.1111/pim.12308
- 40. Jang KS, Han IH, Lee SJ, Yoo J, Kim YS, et al. Experimental rat prostatitis caused by Trichomonas vaginalis infection. Prostate 2019;79(4):379-389. https://doi.org/10.1002/pros.23744
- 41. Bostwick DG. The pathology of benign prostatic hyperplasia. In: Kirby RS, McConnell JD, Fitzpatrick JM, Roehrborn CG, Boyle P, editors. Textbook of Benign Prostate Hyperplasia. 6:2nd ed. Isis Medical Media; London, UK. 2002. p. 97-112.
- 42. De Nunzio C, Salonia A, Gacci M, Ficarra V. Inflammation is a target of medical treatment for lower urinary tract symptoms associated with benign prostatic hyperplasia. World J Urol 2020;38:2771-2779. https://doi.org/10.1007/s00345-020-03106-1
- 43. Gravas S, Cornu JN, Gacci M, Gratzke C, Hermann TRW, et al. Management of Non-Neurogenic Male LUTS. EAU Guidelines Office; Arnhem, the Netherlands. 2022.
- 44. Devlin CM, Simms MS, Maitland NJ. Benign prostatic hyperplasia - what do we know? BJU Int 2021;127(4):389-399. https://doi.org/10.1111/bju.15229
- 45. De Nunzio C, Presicce F, Tubaro A. Inflammatory mediators in the development and progression of benign prostatic hyperplasia. Nat Rev Urol 2016;13(10):613-626. https://doi.org/10.1038/nrurol.2016.168
- 46. Ficarra V, Sekulovic S, Zattoni F, Zazzera M, Novara G. Why and how to evaluate chronic prostatic inflammation. Eur Urol Suppl 2013;12(5):110-115. https://doi.org/10.1016/j.eursup.2013.08.002
- 47. De Marzo AM, Platz EA, Sutcliffe S, Xu J, Gronberg H, et al. Inflammation in prostate carcinogenesis. Nat Rev Cancer 2007;7(4):256-269. https://doi.org/10.1038/nrc2090
- 48. Breyer BN, Huang WY, Rabkin CS, Alderete JF, Pakpahan R, et al. Sexually transmitted infections, benign prostatic hyperplasia and lower urinary tract symptom-related outcomes: results from the prostate, lung, colorectal and ovarian cancer screening trial. BJU Int 2016;117(1):145-154. https://doi.org/10.1111/bju.13050
- 49. Twu O, Dessi D, Vu A, Mercer F, Stevens GC, et al. Trichomonas vaginalis homolog of macrophage migration inhibitory factor induces prostate cell growth, invasiveness, and inflammatory responses. Proc Natl Acad Sci U S A 2014;111(22):8179-8184. https://doi.org/10.1073/pnas.1321884111
- 50. Claus S, Wrenger M, Senge T, Schulze H. Immunohistochemical determination of age related proliferation rates in normal and benign hyperplastic human prostates. Urol Res 1993;21(5):305-308. https://doi.org/10.1007/BF00296825
- 51. Ou Z, He Y, Qi L, Zu X, Wu L, et al. Infiltrating mast cells enhance benign prostatic hyperplasia through IL-6/STAT3/Cyclin D1 signals. Oncotarget 2017;8(35):59156-59164. https://doi.org/10.18632/oncotarget.19465
- 52. Romih R, Winder M, Lee G. Recent advances in the biology of the urothelium and applications for urinary bladder dysfunction. Biomed Res Int 2014;2014:341787. https://doi.org/10.1155/2014/341787
- 53. Pattabiraman G, Bell-Cohn AJ, Murphy SF, Mazur DJ, Schaeffer AJ, et al. Mast cell function in prostate inflammation, fibrosis, and smooth muscle cell dysfunction. Am J Physiol Renal Physiol 2021;321(4):F466-F479. https://doi.org/10.1152/ajprenal.00116.2021
- 54. Kobayashi TK, Fujimoto T, Okamoto H, Yuasa M, Sawaragi I. Association of mast cells with vaginal trichomoniasis in endocervical smears. Acta cytol 1983;27(2):133-137.
- 55. Im SJ, Ahn MH, Han IH, Song HO, Kim YS, et al. Histamine and TNF-alpha release by rat peritoneal mast cells stimulated with Trichomonas vaginalis. Parasite 2011;18(1):49-55. https://doi.org/10.1051/parasite/2011181049
- 56. Han IH, Park SJ, Ahn MH, Ryu JS. Involvement of mast cells in inflammation induced by Trichomonas vaginalis via crosstalk with vaginal epithelial cells. Parasite Immunol 2012;34(1):8-14. https://doi.org/10.1111/j.1365-3024.2011.01338.x
- 57. Beghdadi W, Madjene LC, Benhamou M, Charles N, Gautier G, et al. Mast cells as cellular sensors in inflammation and immunity. Front Immunol 2011;2:37. https://doi.org/10.3389/fimmu.2011.00037
- 58. Krystel-Whittemore M, Dileepan KN, Wood JG. Mast cell: a multi-functional master cell. Front Immunol 2015;6:620. https://doi.org/10.3389/fimmu.2015.00620
- 59. Overed-Sayer C, Rapley L, Mustelin T, Clarke DL. Are mast cells instrumental for fibrotic diseases? Front Pharmacol 2014;4:174. https://doi.org/10.3389/fphar.2013.00174
- 60. Kim SS, Kim JH, Han IH, Ahn MH, Ryu JS. Inflammatory responses in a benign prostatic hyperplasia epithelial cell line (BPH-1) infected with Trichomonas vaginalis. Korean J Parasitol 2016;54(2):123-132. https://doi.org/10.3347/kjp.2016.54.2.123
- 61. Kim JH, Kim SS, Han IH, Sim S, Ahn MH, et al. Proliferation of prostate stromal cell induced by benign prostatic hyperplasia epithelial cell stimulated with Trichomonas vaginalis via crosstalk with mast cell. Prostate 2016;76(15):1431-1444. https://doi.org/10.1002/pros.23227
- 62. Duchesne E, Tremblay MH, Cote CH. Mast cell tryptase stimulates myoblast proliferation; a mechanism relying on protease-activated receptor-2 and cyclooxygenase-2. BMC Musculoskelet Disord 2011;12:235. https://doi.org/10.1186/1471-2474-12-235
- 63. Bagher M, Larsson-Callerfelt AK, Rosmark O, Hallgren O, Bjermer L, et al. Mast cells and mast cell tryptase enhance migration of human lung fibroblasts through protease-activated receptor 2. Cell Commun Signal 2018;16(1):59. https://doi.org/10.1186/s12964-018-0269-3
- 64. Murray DB, McLarty-Williams J, Nagalla KT, Janicki JS. Tryptase activates isolated adult cardiac fibroblasts via protease activated receptor-2 (PAR-2). J Cell Commun Signal 2012;6(1):45-51. https://doi.org/10.1007/s12079-011-0146-y
- 65. Borensztajn K, Bresser P, van der Loos C, Bot I, van den Blink B, et al. Protease-activated receptor-2 induces myofibroblast differentiation and tissue factor up-regulation during bleomycin-induced lung injury: potential role in pulmonary fibrosis. Am J Pathol 2010;177(6):2753-2764. https://doi.org/10.2353/ajpath.2010.091107
- 66. Roman K, Murphy SF, Done JD, McKenna KE, Schaeffer AJ, et al. Role of PAR2 in the development of lower urinary tract dysfunction. J Urol 2016;196(2):588-598. https://doi.org/10.1016/j.juro.2016.01.106
- 67. Noh CS, Chung HY, Han IH, Kim JH, Kim YM, et al. Mast cell tryptase-PAR2 pathway in proliferation of prostatic stromal cells reacted with Trichomonas vaginalis. Parasite Immunol 2021;43(8):e12868. https://doi.org/10.1111/pim.12868
- 68. Lokeshwar SD, Harper BT, Webb E, Jordan A, Dykes TA, et al. Epidemiology and treatment modalities for the management of benign prostatic hyperplasia. Transl Androl Urol 2019;8(5):529-539. https://doi.org/10.21037/tau.2019.10.01
- 69. Kogan-Sakin I, Cohen M, Paland N, Madar S, Solomon H, et al. Prostate stromal cells produce CXCL-1, CXCL-2, CXCL-3 and IL-8 in response to epithelia-secreted IL-1. Carcinogenesis 2009;30(4):698-705. https://doi.org/10.1093/carcin/bgp043
- 70. Siejka A, Schally AV, Barabutis N. The effect of LHRH antagonist cetrorelix in crossover conditioned media from epithelial (BPH-1) and stromal (WPMY-1) prostate cells. Horm Metab Res 2014;46(1):21-26. https://doi.org/10.1055/s-0033-1349127
- 71. Kim JH, Han IH, Kim YS, Noh CS, Ryu JS. Proliferation of prostate epithelia induced by IL-6 from stroma reacted with Trichomonas vaginalis. Parasite Immunol 2018;40(6):e12531. https://doi.org/10.1111/pim.12531
- 72. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014;15:178-196. https://doi.org/10.1038/nrm3758
- 73. Han IH, Kim JH, Jang KS, Ryu JS. Inflammatory mediators of prostate epithelial cells stimulated with Trichomonas vaginalis promote proliferative and invasive properties of prostate cancer cells. Prostate 2019;79(10):1133-1146. https://doi.org/10.1002/pros.23826
- 74. Alonso-Magdalena P, Brossner C, Reiner A, Cheng G, Sugiyama N, et al. A role for epithelial-mesenchymal transition in the etiology of benign prostatic hyperplasia. Proc Natl Acad Sci U S A 2009;106(8):2859-2863. https://doi.org/10.1073/pnas.0812666106
- 75. Shi X, Peng Y, Du X, Liu H, Klocker H, et al. Estradiol promotes epithelial-to-mesenchymal transition in human benign prostatic epithelial cells. Prostate 2017;77(14):1424-1437. https://doi.org/10.1002/pros.23404
- 76. Hu S, Yu W, Lv TJ, Chang CS, Li X, et al. Evidence of TGF-B1 mediated epithelial-mesenchymal transition in immortalized benign prostatic hyperplasia cells. Mol Mem Biol 2014;31(2–3):103-110. https://doi.org/10.3109/09687688.2014.894211
- 77. Jung JH, Ahn SV, Song JM, Chang SJ, Kim KJ, et al. Obesity as a risk factor for prostatic enlargement: a retrospective cohort study in Korea. Int Neurourol J 2016;20(4):321-328. https://doi.org/10.5213/inj.1632584.292
- 78. Lee RK, Chung D, Chughtai B, Te AE, Kaplan SA. Central obesity as measured by waist circumference is predictive of severity of lower urinary tract symptoms. BJU Int 2012;110(4):540-545. https://doi.org/10.1111/j.1464-410X.2011.10819.x
- 79. Sieminska L, Borowski A, Marek B, Nowak M, Kajdaniuk D, et al. Serum concentrations of adipokines in men with prostate cancer and benign prostate hyperplasia. Endokrynol Pol 2018;69(2):120-127. https://doi.org/10.5603/EP.a2018.0006
- 80. Margetic S, Gazzola C, Pegg GG, Hill RA. Leptin: a review of its peripheral actions and interactions. Int J Obes Relat Metab Disord 2002;26:1407-1433. https://doi.org/10.1038/sj.ijo.0802142
- 81. Szyszka M, Tyczewska M, Milecka P, Jopek K, Celichowski P, et al. Effects of leptin on leptin receptor isoform expression and proliferative activity in human normal prostate and prostate cancer cell lines. Oncol Rep 2018;39(1):182-192. https://doi.org/10.3892/or.2017.6066
- 82. Habib CN, Al-Abd AM, Tolba MF, Khalifa AE, Khedr A, et al. Leptin influences estrogen metabolism and accelerates prostate cell proliferation. Life Sci 2015;121:10-15. https://doi.org/10.1016/j.lfs.2014.11.007
- 83. Kim JH, Han IH, Shin SJ, Park SY, Chung HY, et al. Signaling role of adipocyte leptin in prostate cell proliferation induced by Trichomonas vaginalis. Korean J Parasitol 2021;59(3):235-249. https://doi.org/10.3347/kjp.2021.59.3.235