Abstract
The present study intended to verify activities of cysteine proteinase of Pneumocystis carinii from rats and to purify the enzyme. In order to exclude the contamination of host-derived enzymes, concentrates of P. carinii was primarily treated with a mixture of proteinase inhibitors before lysis of P. carinii. A 68-kDa cysteine proteinase was finally purified from the crude extract of P. carinii by 4 sequential chromatographic methods. The enzyme showed an optimal activity at pH 5.5 in 0.1 M sodium acetate, and its activity was specifically inhibited by L-trans-epoxysuccinylleucylamido (4-guanidino) butane (E-64) and iodoacetic acid, suggesting that the enzyme is a cysteine proteinase. The 68-kDa proteinase weakly digested macromolecules such as collagen, hemoglobin and fibronectin. The present study demonstrated the activity of cysteine proteinase at the 68-kDa band of P. carinii, and purified and characterized the molecule.
-
Key words: Pneumocystis carinii, cysteine proteinase, 68-kDa
INTRODUCTION
Pneumocystis carinii is a pathogen with marked fungal genomic affinities, and causes life-threatening pneumonia in immunocompromised patients if untreated (
Walzer, 1994).
Pneumocystis carinii pneumonia (PCP) is one of the most important causes of morbidity and mortality in AIDS patients, and its incidence is growing in individuals submitted to immunosuppressive treatment for cancer, organ transplantation, or chronic inflammatory diseases. In Korea, most people are exposed to
P. carinii within two years after birth (
Hong, 1991), and several human cases of PCP had been reported (
Cheong et al., 1983).
The activity of cysteine proteinases have been known to increase in smokers or lung disease states such as pulmonary granulomatous inflammation and acute lung injury (
Scharfman et al., 1980;
Lesser et al., 1989). There are a few reports on cysteine proteinase activities in
P. carinii infection (
Hayes et al., 1991;
Sukura et al., 1995). In rats with experimentally induced PCP, cathepsin H-like activity was greater than control levels and showed a close relation with parasite burden (
Hayes et al., 1991). Higher collagenase activity was observed in rats with PCP, which returned to normal after the cessation of dexamethasone treatment (
Sukura et al., 1995). Therefore, the activity of cysteine proteinases has been suggested to be closely related with pathogenesis or parasite burden in PCP. However, it was not clearly determined whether the source of the proteinase activity was the host or the parasite.
In an attempt to identify the enzyme activity of P. carinii, the organisms were isolated from the lungs of experimentally infected Wistar rats and the 68-kDa molecule of cysteine proteinase activity was purified and characterized in the present study.
MATERIALS AND METHODS
Induction of PCP in rats
Albino rats of Wistar strain, 7-wk-old, were immunosuppressed by weekly subcutaneous injection of 2 mg methylprednisolone (Depomedrol®, Upjohn Korea Co.) for 8-12 wk. The rats were kept in conventional animal rooms with regular diet and tap water including ampicillin in a dosage of 1 mg/ml tap water.
Isolation and preparation of the crude extract of P. carinii
The lungs were removed after sacrifice of rats by cervical dislocation under the ether anesthesia at 8 wk of immunosuppression. The impression smears were made with five cut surfaces of the lungs, and stained with modified Giemsa solution (Diff-Quik®, Fisher Scientific Co., USA) for observation of P. carinii. The lungs infected with bacteria or fungi were discarded.
Pneumocystis carinii were isolated and pooled by the modified method of Hong et al (
1994). Briefly, host tissues and cells were filtered through the cotton gauze after chopping and homogenizing the lungs. The remaining host blood cells and pneumocytes were lysed by successive treatment in 0.05 M ammonium chloride and hypotonic solution. The homogenate was treated with Complete™ protease inhibitor (Boehringer Mannheim, Germany), washed three times by centrifugation at 2,500
g at 4℃ for 10 minutes, and then filtered through 10 µm pore membrane. After one more treatment with Complete™ protease inhibitor and successive washings, the mass of
P. carinii was sonificated using sonicator (Heat Systems-Ultrasonic Inc., New York, USA). The supernatant was regarded as the crude extract of
P. carinii after centrifugation at 10,000
g at 4℃ for 1 hr. The homogenate of healthy control rat lungs was also prepared as above, and used as the control.
The purity of P. carinii isolation was determined by microscopic observation of the concentrate before sonification during the isolation procedure. In order to exclude the possible contamination of any proteins derived from the host tissue, the crude extract of P. carinii and the homogenate of normal rat lung were examined through SDS-PAGE running.
Proteolytic assay
The enzyme activity was assayed using carboxybenzoyl-phenylalanyl-arginyl-7-amino-4-methylcoumarin (CBZ-phe-arg-AMC) as a fluorogenic substrate (Sigma). The reaction mixtures were composed of 440 µl phosphate buffered saline (PBS; 0.1 M, pH 7.0), 20 µl enzyme solution, 10 µl substrate, and 5 µl 0.1 M dithiothreitol (DTT), which were incubated for 2 hr at 37℃. The proteolytic activity was measured by a DNA fluorometer TKO 100 (excitation wavelength = 380 nm, emission wavelength = 460 nm) (Hoefer, San Francisco, USA) after the termination of reaction by addition of 10 µl iodoacetic acid (IAA) and 400 µl 7.2% ZnSO4. The amount that released 1 µM AMC for 1 hr was regarded as one unit of the enzyme activity.
Purification of the enzyme
The crude extract of P. carinii was applied to a DEAE-sepharose fast flow column (Pharmacia, Piscataway, USA), 1.6 × 4.0 cm long, pre-equilibrated with 25 mM Tris-HCl (pH 7.4). Proteins were eluted with the same buffer containing 0, 0.05, 0.1, and 0.2 M NaCl at a flow rate of 40 ml/hr. Fractions showing high enzyme activities were pooled, dialysed, lyophilized, and reconstituted with 500 µl sodium acetate buffer (20 mM, pH 6.4) for Q-sepharose chromatography.
A total of 450 µl of enzyme solution was challenged to a Q-sepharose column (Pharmacia), 1.6 × 4.0 cm long, pre-equilibrated with 20 mM sodium acetate buffer (pH 6.4). The elution of proteins was carried out with the same buffer containing 0, 0.05, 0.1, and 0.2 M NaCl at a flow rate of 40 ml/hr. Enzymatically active fractions were pooled, dialysed, lyophilized, and reconstituted with 500 µl 25 mM Tris-HCl (pH 7.4) for final purification.
The enzyme was finally purified after arginine-sepharose 4B chromatography (Pharmacia). Proteins were fractionated by the application of NaCl solution in a stepwise fashion at a flow rate of 40 ml/hr.
SDS-PAGE was done for observation of a purified protein using a 7.5-15% separating gel. The standard molecular weight markers were phosphorylase B (94 kDa), albumin (67), ovalbumin (43), carbonic anhydrase (30), trypsin inhibitor (20.1), and α-lactalbumin (14.4) (Pharmacia).
Biochemical characteristics
Twenty microliters of the purified enzyme was incubated in 0.01, 0.05, 0.1, 0.2, 0.3, 0.4 and 0.5 M PBS (440 µl each, pH 7.0) for 3 hr at 37℃. The optimal pH was also measured in sodium acetate (0.1 M) at pH 4, 4.5, 5, and 5.5, in sodium phosphate (0.1 M) at pH 6, 6.5, 7, and 7.5, and in Tris-HCl (0.1 M) at pH 8.0, respectively.
Modulation effects on enzyme activity were monitored by incubation of 10 µl enzyme with various inhibitors at room temperature for 20 min, and then 20 µl DTT and 10 µl substrate were added to the reaction mixture. Effectors used in the present study were DTT (5 mM), L-trans-epoxy-succinylleucylamido (4-guanidino) butane (E-64, 0.01 mM), IAA (1 mM), leupeptin (0.1 mM), 4-(amidinophenyl) methanesulfonyl fluoride (APMSF, 0.1 mM), aprotinin (10 µg/ml), ethylene diamine tetraacetate (EDTA, 2mM) and 1, 10-phenanthroline (0.1 mM) (Sigma).
Enzyme activities against macromolecules
Three kinds of macromolecules were incubated with the enzyme to determine the digestive activity of the enzyme. They were acid soluble calf skin collagen (type I, (Boehringer Mannheim), hemoglobin (Sigma), and fibronectin isolated from human plasma. Reaction mixtures containing 20 µl diluted enzyme (8 µg of protein) and macromolecular substrates were incubated for 1, 3, 5, and 12 hr at 37℃. The amounts of collagen, hemoglobin, and fibronectin added to the mixtures were 17 µl (80 µg), 13 µl (65 µg), and 21 µl (80 µg), respectively. Digested products were visualized by SDS-PAGE.
RESULTS
Pneumocystis carinii were collected from the rat lungs with severe infection, which was mainly composed of trophic forms. The lung smears and preparation of
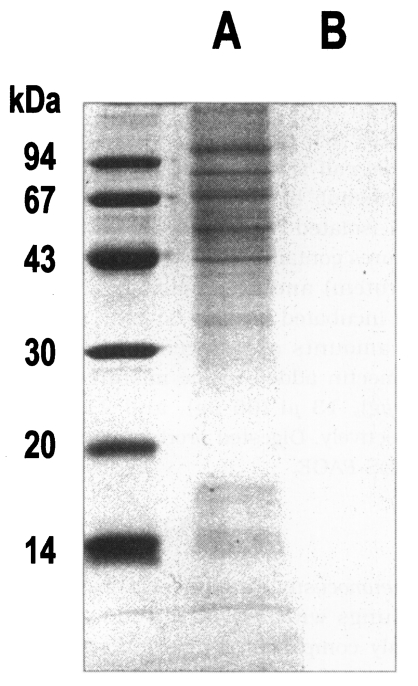
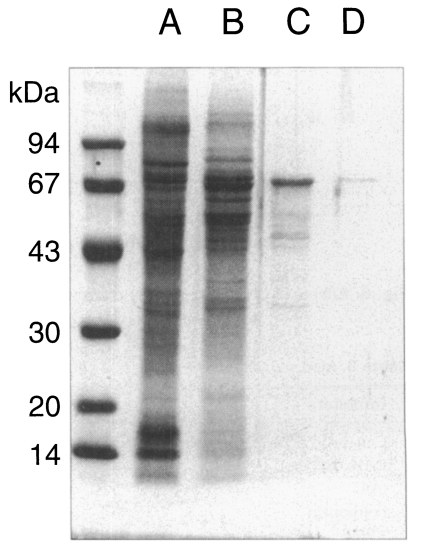
P. carinii were free of bacterial and fungal infection at the light-microscope level. Although the purity of isolation was determined as 99%, it was not sure whether the preparation was totally free of contamination of host cells. However, the homogenate of normal rat lung showed no protein bands at all, while the crude extract of
P. carinii revealed many bands by SDS-PAGE (
Fig. 1). The homogenate of the normal rat lung showed an endogenous proteolytic activity of one thrid or a half level of that of
P. carinii extracts, but exhibited negligible enzyme activity when treated with a mixture of protease inhibitors during the same procedure of
P. carinii isolation (
Table 1). Therefore, it was confirmed that the proteolytic activity of
P. carinii extract was derived from
P. carinii only without the effect of host cells.
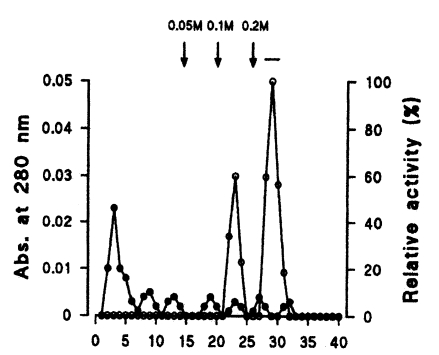
Seven protein peaks were obtained by applying the crude extract to a DEAE-sepharose fast flow column, of which two peaks exhibited enzyme activities (
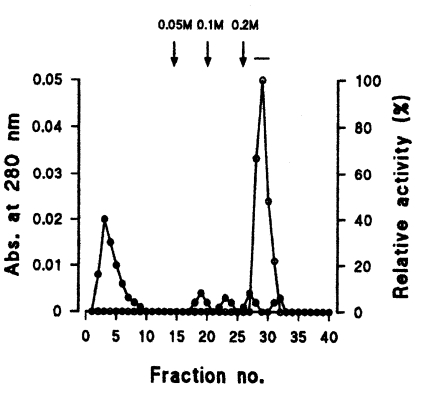
Fig. 2). The 0.2 M NaCl fraction showing highest proteolytic activity was further purified by Q-sepharose chromatography. Among five protein peaks, the enzyme activity was observed only in the 0.2 M NaCl fraction (
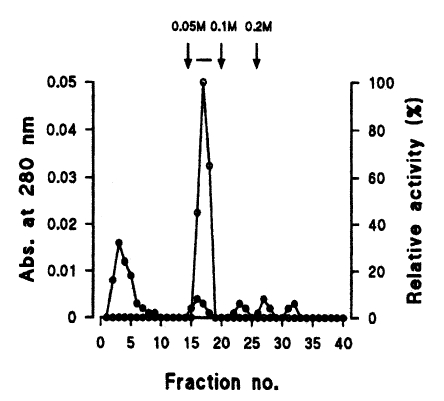
Fig. 3). Finally, a single active protein band was obtained in the 0.05 M NaCl fraction by arginine-sepharose 4B chromatography (
Fig. 4), and its molecular weight was 68 kDa (
Fig. 5).
The activities and yields of the enzyme during purification steps are summarized in
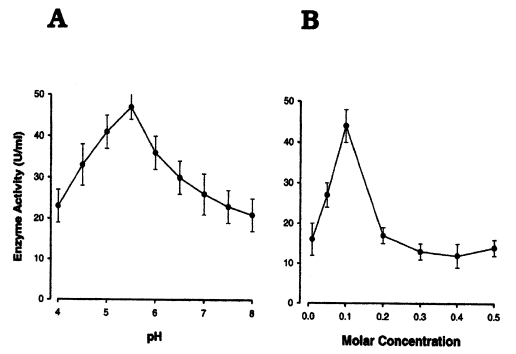
Table 2. The activity of the purified enzyme was optimal in 0.1 M of buffer, and was the highest at pH 5.5 (
Fig. 6A, 6B).
Proteolytic activities were potentiated two times by the addition of 5 mM DTT into the reaction mixtures (
Table 3), and therefore, the modulatory effect of proteinase inhibitors was determined in the presence of DTT. Cysteine proteinase specific inhibitors, IAA and E-64, strongly inhibited the proteolytic activity of the purified enzyme, and a common inhibitor of both cysteine and serine proteinase, leupeptin, also showed a suppressive effect on enzyme activity (
Table 3). On the contrary, serine proteinase specific inhibitors, aprotinin and APMSF, and metallo-proteinase inhibitors, EDTA and 1,10-phenanthroline had no inhibitory effect on the enzyme activity. The 68-kDa enzyme weakly degraded macromolecules such as collagen type I, hemoglobin and fibronectin (data not shown).
DISCUSSION
In the present study, cysteine proteinase activity was observed and a 68-kDa proteinase was finally purified from the crude extract of P. carinii by sequential separation processes of DEAE-sepharose fast flow, Q-sepharose, and arginine-sepharose 4B chromatographies. The activity of the purified enzyme was the highest at pH 5.5 with 0.1 M sodium acetate, and enhanced in the presence of 5 mM DTT. The enzyme was determined to be an acidic cysteine proteinase because of its acid optimum pH and specific inhibition by cysteine proteinase inhibitors, E-64 and IAA.
Biochemical contamination of the crude extract of
P. carinii with that of hosts or other microbial agents is critical in the present study. Since in vitro culture systems for
P. carinii are not still available, an animal model using Wistar rats was used in this study (
Atzori et al., 1998;
Dei-Cas et al., 1998). Therefore the biochemical purity of
P. carinii crude extract should be evaluated. In the present study, host tissues or pneumocytes were eliminated by filtration through 10 µm pore membrane and remaining cells were lysed in hypotonic solution. By the process, trophic forms of
P. carinii were isolated with a high purity, which was confirmed by microscopic observation of the smears. Also the cocktail of proteinase inhibitors could exclude the possibility of contamination by host enzymes of cysteine proteinase activity.
Enzymatic assay and SDS-PAGE demonstrated that the crude extract showed a proteolytic activity derived from P. carinii only, whereas the normal rat lung homogenate treated with inhibitors showed no activity. Therefore, it was confirmed that the crude extract of P. carinii had its own enzyme activity alone with no contamination of host enzymes.
Several investigators observed the enzyme activities of
P. carinii in the bronchoalveolar lavage fluid or the lung homogenate. Among three kinds of cysteine proteinases, cathepsin B-, and cathepsin L-like activities of lung homogenate were found to increase from 8 week of immunosuppressive treatment, while cathepsin H-like activity was greater than control levels from 3 week of immunosuppression, and closely correlated with the parasite burden (
Hayes et al., 1991). Activities of all of the three enzymes were also detected in lysates of purified
P. carinii, however, each proteinase responsible for the observed activities was not purified biochemically. Collagenase and serine proteinase activities were found in the bronchoalveolar lavage fluid of rats with PCP, which may lead to damage of the basal lamina and interstitial connective tissue and thereby can contribute to impaired alveolar function during PCP (
Sukura et al., 1995). Carboxypeptidase M activity has been demonstrated to be elevated in the bronchoalveolar lavage fluid of patients with acute lung disease such as acute pneumonia, PCP, and lung cancer, which may be an indicator of type I cell injury in lung diseases (
Dragovic et al., 1995). Other host factors are thought to be involved in the pathogenesis of PCP. The primary injury in PCP is the increased permeability of the alveolar capillary membrane, which may be produced by the host response against
P. carinii (
Sukura et al., 1995). Urokinase-type plasminogen activator, which is a host cell origin proteinase intimately involved in inflammatory cell recruitment and activation, is also an important enzyme in development of PCP (
Beck et al., 1999). The knockout mice of the proteinase gene were heavily infected while wild type mice were almost free from the infection. Host and parasite factors may together play their roles in the pathogenesis of PCP.
The subtilisin-like serine proteinases of
P. carinii was confirmed and its gene has been cloned (
Lugli et al. 1997;
Wada and Nakamura, 1997). The proteinase may be involved in cleavage of major surface glycoproteins or other related proteins of
P. carinii (
Wada and Nakamura, 1997). A conserved N-terminal portion of major surface glycoproteins has been suggested to be processed by subtilisin-like proteinases, and a variant portion of major surface glycoproteins may be exposed on the cell surface of
P. carinii for host immune evasion (
Wada and Nakamura, 1997). However, the purification of this subtilisin-like proteinase is highly needed for further evaluation of its roles in the pathogenesis of PCP.
The purified enzyme molecule of 68-kDa in the present study showed weak degradative activities against macromolecules such as collagen, hemoglobin and fibronectin. The function of the 68-kDa cysteine proteinase of P. carinii is still poorly understood and it is a topic of further study.
Notes
-
This study was supported by the financial support of the Korea Research Foundation made in the program year of 1998 (1998-021-F00182) and the Ministry of Education, Korea (1997).
ACKNOWLEDGEMENTS
The authors are grateful to Miss Yeon Hee Lim, Department of Parasitology, Seoul National University College of Medicine, for her technical assistance in the enzyme purification.
References
- 1. Atzori C, Aliouat EM, Bartlett MS, Dujardin L, Cargnel A, Del-Cas E. Current in vitro culture systems for Pneumocystis. FEMS Immunol Med Microbiol 1998;22:169-172.
- 2. Beck JM, Preston AM, Gyetko MR. Urokinase-type plasminogen activator in inflammatory cell recruitment and host defense against Pneumocystis carinii in mice. Infect Immun 1999;67:879-884.
- 3. Cheong SK, Im CB, Ahn DH, Sohn KC. Pneumocystis carinii pneumonia in an institution for adoption. J Korean Med Assoc 1983;26:329-336. (in Korean).
- 4. Dei-Cas E, Brun-Pascaud M, Bille-Hansen V, Allaert A, Aliouat EM. Animal models of pneumocystosis. FEMS Immunol Med Microbiol 1998;22:163-168.
- 5. Dragovic T, Schraufnagel DE, Becker RP, Sekosan M, Votta-Velis EG, Erdos EG. Carboxypeptidase M activity is increased in bronchoalveolar lavage in human lung disease. Am J Respir Crit Care Med 1995;152:760-764.
- 6. Hayes DJ, Stubberfield CR, McBride JD, Wilson DL. Alterations in cysteine proteinase content of rat lung associated with development of Pneumocystis carinii infection. Infect Immun 1991;59:3581-3588.
- 7. Hong ST. Serologic responses to Pneumocystis carinii of Seoul National University Hospital patients. Korean J Parasitol 1991;29:355-361.
- 8. Hong ST, Yu JS, Lee M. Relation of cyst counts with numbers of total nuclei of Pneumocystis carinii in rats. Korean J Parasitol 1994;32:171-176.
- 9. Lesser M, Chang JC, Galicki NI, Edelman J, Cardozo C. Cathepsin B and D activity in alveolar macrophages from rats with pulmonary granulomatous inflammation or acute lung injury. Agents Actions 1989;28:264-271.
- 10. Lugli EB, Allen AG, Wakefield AE. A Pneumocystis carinii multi-gene family with homology to subtilisin-like serine proteases. Microbiology 1997;143:2223-2236.
- 11. Scharfman A, Lafitte JJ, Tunnel AB, Aerts C, Sablonniere B, Roussel P. Glycosidases and proteases of alveolar macrophages obtained by bronchoalveolar lavage from smokers and non-smokers. Lung 1980;157:135-142.
- 12. Sukura A, Konttinen YT, Sepper R, Kaartinen L, Sorsa T, Lindberg L. Collagenases and the serine proteinases elastase and cathepsin G in steroid-induced Pneumocystis carinii pneumonia. J Clin Microbiol 1995;33:829-834.
- 13. Wada M, Nakamura Y. cDNA cloning and overexpression of cell surface subtilisin-like proteases (SSP) of Pneumocystis carinii. J Euk Microbiol 1997;44:54S.
- 14. Walzer PD. Pneumocystis carinii pneumonia. Lung biology in health and disease. 1994. Vol. 69:2nd ed. New York, USA. Marcel Dekker, Inc.
Fig. 1SDS-PAGE analysis of crude extract of
Pneumocystis carinii (A) and homogenate of
normal rat lung (B).
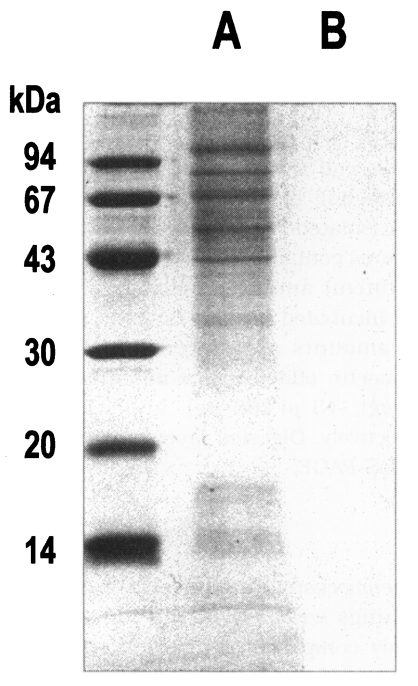
Fig. 2Elution profiles of a protease by DEAE-sepharose fast flow chromatography. Fractions were assayed for activity on CBZ-phe-arg-MNA (○) and monitored for protein content (●) at 280 nm. Fractions with high enzyme activities were pooled, as shown by the bar (-).

Fig. 3Elution profiles of a protease by Q-sepharose chromatography. Fractions were assayed for activity on CBZ-phe-arg-MNA (○) and monitored for protein content (●) at 280 nm. Fractions with high enzyme activities were pooled, as shown by the bar (-).
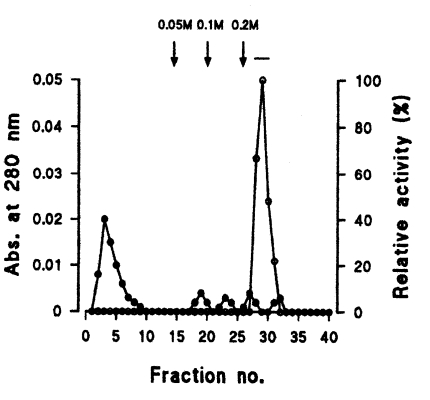
Fig. 4Elution profiles of a protease by arginine-sepharose 4B chromatography. Fractions were assayed for activity on CBZ-phe-arg-MNA (○) and monitored for protein content (●) at 280 nm. Fractions with high enzyme activities were pooled, as shown by the bar (-).

Fig. 5SDS-PAGE analysis of proteins purified from crude extract to a cysteine protease by sequential chromatographic steps. A, crude extract; B, 0.2 M NaCl fraction from DEAE-sepharose fast flow; C, 0.2 M NaCl fraction from Q-sepharose; D, 0.05 M NaCl fraction from arginine-sepharose 4B.

Fig. 6Effects of pH and molar concentration on a cysteine proteinase of Pneumocystis carinii.
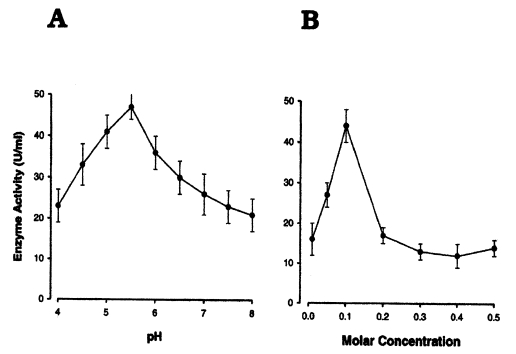
Table 1.Proteolytic activities of four kinds of homogenates of the rat lungs or Pneumocystis carinii
Table 1.
|
Homogenates |
Activity (unit/ml) |
Specific activity (unit/mg) |
|
Normal rat lung |
21.5 |
5.2 |
|
Normal rat lung + inhibitor |
2.0 |
0.4 |
|
P. carinii
|
18.0 |
13.9 |
|
P. carinii + inhibitor |
13.5 |
16.9 |
Table 2.Summary of purification from the crude extract to a proteinase of Pneumocystis carinii
Table 2.
|
Step |
Total protein (mg) |
Total activity (units) |
Specific activity (units/mg) |
Purification (fold) |
Recovery (%) |
|
Crude extract |
29.6 |
3031.5 |
102.4 |
1.0 |
100 |
|
DEAE-sepharose fast flow |
3.5 |
1730.1 |
494.3 |
4.8 |
57.1 |
|
Q-sepharose |
2.1 |
1252.0 |
596.2 |
5.8 |
41.3 |
|
Arginine-sepharose 4B |
0.8 |
755.6 |
944.5 |
9.2 |
24.9 |
Table 3.Modulatory effects of various effectors on the enzyme activity
Table 3.
|
Inhibitors |
Final concentration |
Relative activitya) (%) |
|
Control (DTT-activated) without DTT |
5 mM |
100.0 ± 7.5 |
|
E-64 |
10 μM |
0.4 ± 0.2 |
|
IAA |
1 mM |
0.5 ± 0.3 |
|
Leupeptin |
0.1 mM |
9.1 ± 3.8 |
|
APMSF |
0.1 mM |
99.9 ± 3.8 |
|
Aprotinin |
10 μg/ml |
107.0 ± 3.7 |
|
EDTA |
2 mM |
103.4 ± 2.5 |
|
1,10-phenanthroline |
5 mM |
97.9 ± 7.1 |