Abstract
Identification of the genes responsible for the recovery of virulence in brain-passaged Acanthamoeba culbertsoni was attempted via mRNA differential display-polymerase chain reaction (mRNA DD-PCR) analysis. In order to identify the regulatory changes in transcription of the virulence related genes by the brain passages, mRNA DD-PCR was performed which enabled the display of differentially transcribed mRNAs after the brain passages. Through mRNA DD-PCR analysis, 96 brain-passaged amoeba specific amplicons were observed and were screened to identify the amplicons that failed to amplify in the non-brain-passaged amoeba mRNAs. Out of the 96 brain-passaged amoeba specific amplicons, 12 turned out to be amplified only from the brain-passaged amoeba mRNAs by DNA slot blot hybridization. The clone, A289C, amplified with an arbitrary primer of UBC #289 and the oligo dT11-C primer, revealed the highest homology (49.8%) to the amino acid sequences of UPD-galactose lipid transferase of Erwinia amylovora, which is known to act as an important virulence factor. The deduced amino acid sequences of an insert DNA in clone A289C were also revealed to be similar to cpsD, which is the essential gene for the expression of type III capsule in group B streptococcus. Upregulated expression of clone A289C was verified by RNA slot blot hybridization. Similar hydrophobicity values were also observed between A289C (at residues 47-66) and the AmsG gene of E. amylovora (at residues 286-305: transmembrane domains). This result suggested that the insert of clone A289C might play the same function as galactosyl transferase controlled by the AmsG gene in E. amylovora.
-
Key words: free-living amoebae, Acanthamoeba culbertsoni, virulence, mRNA DD-PCR analysis, slot blot hybridization
INTRODUCTION
Free-living amoebas of
Acanthamoeba sp. induce granulomatous amoebic encephalitis and keratitis (
Cohen et al., 1985,
1987;
Ishibashi et al., 1987;
Stehr-Green et al., 1987). The virulence of the pathogenic amoebas can be decreased by a long-term in vitro cultivation, and it can be recovered by intranasal infection or mouse brain passages (
Lee et al., 1983;
Mazur and Hadas, 1994). On the basis of the recovery of virulence, it may be postulated that the secretion of some biochemical products related to the virulence is increased. For example, the presence of proteinase was confirmed in pathogenic
Acanthamoeba (
Hadas and Mazur, 1993) and the increased activities of peroxidase and proteinase were also observed in
Acanthamoeba as the virulence was recovered by mouse brain passages (
Mazur and Hadas, 1994).
In this study, mouse brain passages were performed with Acanthamoeba culbertsoni that had been maintained in the laboratory for several years in order to monitor any change in the level of the virulence. We tried to identify upregulated mRNAs by mRNA differential display-polymerase chain reaction (mRNA DD-PCR), which was responsible for the increase of virulence in mouse-brain-passaged amoebas.
MATERIALS AND METHODS
Cultivation of amoebas and mouse brain passages
Acanthamoeba culbertsoni isolated at the Prince Leopold Institute of Tropical Medicine in Belgium was cultured in Casitone, Glucose and Vitamin (CGV) medium at 37℃. Trophozoites of A. culbertsoni (1×106 trophozoites per 5 ml saline) were inoculated intranasally into male ICR mice under anesthesia. Mortality and behavior of the infected ICR mice were monitored 25 days post-inoculation (PI). Thiry-inoculated ICR mice of around 20 g were prepared as a control group. The recovered amoebas were cultured briefly in CGV medium and directly used for the consecutive mouse infection. In order to recover amoebas, autopsy was performed immediately after sacrificing the animals and some pieces of the forebrain tissue were placed in culture medium.
Purification of mRNA and synthesis of single stranded cDNA
Total RNA was extracted by the method of Chomczynski and Sacchi (
1987) and quantified with a Dipstick kit (Invitrogen, San Diego, CA, USA). mRNA was purified from the total RNA by using a magnetic oligo dT
25-bead (Dynabeads®, Oslo, Norway). Three kinds of the first stranded cDNA templates for DD-PCR were synthesized from the purified mRNA by using one of the 3 one-base anchored oligo-dT primers (i.e., 5'-TTTTTTTTTTT-N-3': N is either A, G, or C). Synthesized cDNA templates were quantified using a Dipstick kit and stored at -20℃.
The mixture for mRNA DD-PCR consisted of 30 pg template cDNA, 0.38 µM arbitrary primer (Biotechnology Laboratory, University of British Columbia, Vancouver, Canada), 0.4 µM one-base anchored oligo-dT11 primer which was identical to the one used for cDNA synthesis, 1X reaction buffer (100 mM Tris-HCl, 500 mM KCl, pH 8.3, Boehringer Mannheim, Mannheim, Germany), 250 µM dNTPs, 1.5 mM MgCl2, and 1.5 unit Taq DNA polymerase (Promega, WI, USA). PCR was performed as follows: 40 cycles of denaturation at 94℃ for 5 sec, annealing at 42℃ for 60 sec, primer extension at 72℃ for 90 sec and final extension at 72℃ for 10 min. PCR products (15 µl of 20 µl total volume) were fractionated by 5% polyacrylamide gel electrophoresis with the salt-gradient buffer system. The remaining PCR products (5 µl each) were pooled into two groups, the brain-passaged group (tester group) and the non-brain-passaged group (driver group), to prepare the probes for Southern analysis. By comparing the electrophoretic migration of the PCR products between two groups, PCR products amplified only in the tester sample were selected for reamplification. Unique DNA fragments in the tester were purified from the gel and reamplified using the identical primer set.
DNA slot-blot hybridization
Reamplified PCR products were purified by using a Gene Spin kit (Bio 101, Inc., Vista, CA, USA) and blotted onto a nylon membrane (Tropix, Bedford, MA, USA) using slot-blot filtration manifolds (Hoeffer, San Francisco, CA, USA). Pooled PCR products of both groups were purified with a Gene Spin kit. Purified PCR products were labeled with biotin-14-dCTP by using a random primer biotin DNA labeling kit (Tropix), with the use of a random octamer as a primer. Hybridization reaction was performed with a Southern-light™ chemiluminescent detection system (Tropix). After hybridization, membrane was exposed to standard Kodak XAR X-ray film for 30 min.
Cloning and sequencing analysis
Tester-specific amplicons were ligated to pGEM easy T-vector (Promega) which were then transformed into E. coli strain of DH5α. Transformed cells were screened on an x-gal plate. Positive colonies were selected and plasmids were purified. The DNA insert was confirmed by a restriction enzyme digestion, purified with a wizard plus minipreps kit (Promega), and sequenced. Obtained nucleotide sequences were interpreted to amino acid sequences using the WDNASIS V.2.0 (Hitachi Software Engineering Co., Ltd., Japan). computer program. Homologous amino acid sequence searching was performed by BLAST search. The hydrophobicity between the most similar ones was tested by the method of Kyte and Doolittle using WDNASIS V.2.0. to verify whether they had the identical protein function or not.
RNA slot-blot hybridization
In order to verify if there was differential expression of the brain passaged amoeba-specific mRNA, slot-blot hybridization was performed. RNAs extracted from both brain-passaged and non-brain-passaged amoebas were blotted onto the nylon membrane by slot-blot manifold. They were further processed with slot-blot hybridization as described above.
RESULTS
mRNA DD-PCR was performed with the first stranded cDNAs of the brain-passaged amoeba and the long-term in vitro-cultured amoeba by using 66 pairs of 22 arbitrary primers and 3 oligo-dT
11-N primers. A total of 96 amplicons was observed from the brain-passaged amoeba (
Fig. 1;
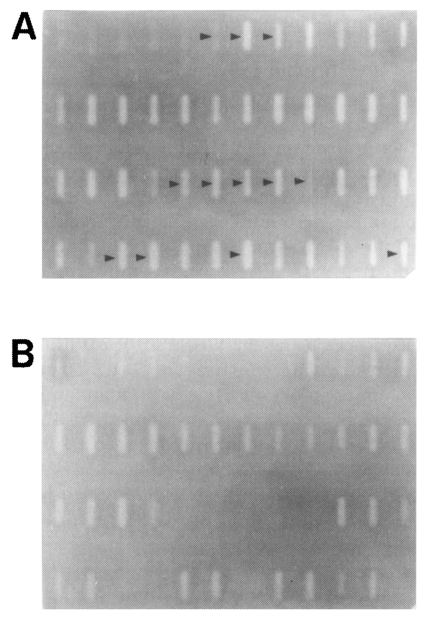
Table 1). Out of these, a total of 12 amplicons was specifically hybridized to the probes prepared from the brain-passaged group, but not to the probes from the non-brain-passaged group (
Fig. 2;
Table 2).
The DNA sequences of the 12 brain-passaged amoeba cDNA were determined and interpreted to amino acid sequences (
Fig. 3). Among them, the interpreted amino acid sequences of an insert DNA (750 bp) in clone A289C, amplified with an arbitrary primer of UBC #289 and oligo dT
11-C primer, revealed the highest homology to the amino acid sequences of UPD-galactose lipid transferase of
Erwinia amylovora. UPD-galactose lipid transferase of
E. amylovora has been known to act as an important virulence factor. The interpreted amino acid sequences of an insert DNA in A289C clone also showed 49.8% similarity and 34.4% identity to the
AmsG gene in
E. amylovora, and they were found to be similar to
cpsD, which is the essential gene for the expression of type III capsule in group B streptococcus (
Fig. 4). Hydrophobicity profiles of clone A289C and the UPD-galactose lipid transferase gene in
E. amylovora (EMBL accession No. Q46628) were investigated by WDNASIS V.2.0. The result showed that overall hydrophobicity profiles of the two proteins were very similar (data not shown). Especially, the transmembrane domain (residues 286-305) of the
AmsG gene in
E. amylovora and its corresponding region (residue 47-66) of clone A289C showed the highest value of hydrophobicity. The result of RNA slot-blot hybridization probed with the insert of clone A289C revealed that the expression of the insert DNA (i.e., a portion of a gene) was upregulated in the brain-passaged amoeba (
Fig. 5).
DISCUSSION
It has been postulated that the expression of genes directly responsible for virulence may be upregulated by consecutive brain passages of
A. culbertsoni. Since the virulence of a long-term cultured
A. culbertsoni in the laboratory is recovered via consecutive brain passages (
Lee et al., 1983;
Mazur and Hadas, 1994), mRNA DD-PCR has been successfully applied to analyze the differences in the gene expression according to different tissues or to different developmental stages by comparing reverse transcribed cDNA from the purified mRNA of different samples. For example, cancer cell-specific mRNA (
Liang et al., 1992), human brain tissue-specific mRNA (
Sokolov and Prockop, 1994), and developmental stage specific mRNA both in
Eimeria bovis of the coccidian parasite (
Abrahamsen et al., 1995) and in
Fasciola hepatica (
Reed et al., 1998) have been successfully identified. This method has also been applied to verify whether there is a differential expression of any genes during excystation of
Giardia lamblia or not (
Hetsko et al., 1998).
The principle of mRNA DD-PCR is that three kinds of cDNA pools are synthesized from mRNA templates by using different oligo-dT11-N (N is either A, G, or C) primers during reverse transcription reaction. Each cDNA pool is analyzed by PCR using the same oligo-dT11-N primer used for cDNA synthesis and an arbitrary 10-mer primer, which enables random amplification of cDNA fragments depending on the homologous sequences. Differences in amplification profiles of the compared samples, which are gauged mainly by the presence or absence of the amplification product of the same size, represents the expressional differences of the genes between the samples. In this study, mRNA DD-PCR was performed to identify the expressional differences of the gene(s) controlling the synthesis of virulence-related biochemical compounds in brain-passaged A. culbertsoni. A total of 96 amplification products was observed in brain-passaged amoeba. Among them, 12 amplicons were verified to be amplified from the brain-passaged amoeba-specific cDNAs which were reverse transcribed from differentially-expressed mRNA by the brain passages. Apart from these 12 brain-passaged amoeba-specific amplification products, the remaining amplification products, although they were observed only in the brain-passaged amoeba, turned out to be the false-positive signals which were also frequently observed in other studies using mRNA DD-PCR technique.
On the basis of sequencing analysis, clone A289C (750 bp amplification product of mRNA DD-PCR with a combination of the arbitrary primer #289 and oligo dT
11-C) was verified as a gene controlling the expression of transferase. By BLAST searching analysis, the most similar amino acid sequence was found to be UPD-galactose lipid transferase of
E. amylovora, which is known to be controlled by the
AmsG gene and acts as an important virulence factor (
Bugert et al., 1995). The amino acid sequence of the clone A289C was also similar to that of the
cpsD gene, which is known to be an important factor for the expression of type III capsule in group B streptococcus. Galactosyl transferase is known to be a prerequisite in the assembly of the group B streptococci type III capsular polysaccharide, which is an important element of the virulence function in the capsule (
Rubens et al., 1993). This finding was supported by the hydrophobicity value at residues 47-66 in the clone A289C, which was identical to the transmembrane domains (residues 286-305) of the
AmsG gene in
E. amylovora. The high similarity of hydro-phobicity, including the transmembrane domain, seems that the clone A289C is closely related or the same as the
AmsG gene both in the structure and the function. RNA slot-blot analysis also revealed that the DNA insert of clone A289C was amplified from upregulatedly-expressed mRNA of the brain-passaged amoeba whose virulence was recovered. Galactosyl transferase is also well-known as an important factor for pathogenesis in several bacteria including salmonella and
Escherichia coli (
Jiang et al., 1991) On the basis of this observation, it was postulated that an insert of the clone A289C might have the same function as galactosyl transferase which is controlled by the
AmsG gene in
E. amylovora. Also, it may play an important role in the recovery of virulence in
A. culbertsoni by brain passages.
Notes
-
This study was supported by the Basic Medicine Research Fund, Ministry of Education, Republic of Korea (1996)
References
- 1. Abrahamsen MS, Johnson RR, Hathaway M, White MW. Identification of Eimeria bovis merozoite cDNAs using differential mRNA display. Mol Biochem Parasitol 1995;71:183-191.
- 2. Bugert P, Geider K. Molecular analysis of the ams operon required for exopolysaccharide synthesis of Erwinia amylovora. Mol Microbiol 1995;15:917-933.
- 3. Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987;162:156.
- 4. Cohen EJ, Buchanan HW, Laughrea PA, et al. Diagnosis and management of Acanthamoeba keratitis. Am J Ophthalmol 1985;100:389-395.
- 5. Cohen EJ, Parlato CJ, Arentsen JJ, et al. Medical and surgical treatment of Acanthamoeba keratitis. Am J Ophthalmol 1987;103:615-625.
- 6. Hadas E, Mazur T. Proteolytic enzymes of pathogenic and non-pathogenic strains of Acanthamoeba spp. Trop Med Parasitol 1993;44:197-200.
- 7. Hetsko ML, McCaffery JM, Svard SG, et al. Cellular and transcriptional changes during excystation of Giardia lamblia in vitro. Exp Parasitol 1998;88:172-183.
- 8. Ishibashi Y, Matsumoto R, Watanabe R, et al. Case of Acanthamoeba keratitis. Acta Soc Ophthalmol Jpn 1987;92:963-972.
- 9. Jiang XM, Neal B, Santiago F, et al. Structure and sequence of the rfb (O antigen) gene cluster of Salmonella serovar typhimurium (strain LT2). Mol Microbiol 1991;5:695-713.
- 10. Lee DK, Lee KT, Im KI. Changes in the pathogenicity of Naegleria fowleri by several brain passaged in mice. Korean J Parasitol 1983;21:234-240.
- 11. Liang P, Averboukh L, Keyomarsi K, Sager R, Pardee AB. Differential display and cloning of mRNAs from human breast cancer versus mammary epithelial cells. Cancer Res 1992;52:6966-6968.
- 12. Mazur T, Hadas E. The effect of the passages of Acanthamoeba strains through mice tissues on their virulence and its biochemical markers. Parasitol Res 1994;80:431-434.
- 13. Reed MB, Spothill TW, Strugnell RA, Panaccio M. Fasiola hepatica: Stage-specific expression of novel gene sequences as identified by differential display. Exp Parasitol 1998;89:169-179.
- 14. Rubens CE, Heggen LM, Haft RF, Wessels MR. Identification of cpsD, a gene essential for type III capsule expression in group B streptococci. Mol Microbiol 1993;8:843-855.
- 15. Sokolov BP, Prockop DJ. A rapid and simple PCR-based method for isolation of cDNA from diffrentially expressed genes. Nucleic Acids Res 1994;22:4009-4015.
- 16. Sompayrac L, Jane S, Burn TC, Tenen DG, Danna KJ. Overcoming limitation of the mRNA differential display technique. Nucleic Acids Res 1995;23:4738-4739.
- 17. Stehr-Green JK, Bailey TM, Visvesvara GS. Acanthamoeba keratitis in soft contact lens wearers. A case control study. JAMA 1987;258:57-60.
Fig. 1An example of 5% polyacrylamide gel electrophoresis with DDRT-PCR products. PCR was performed with combinations of an arbitrary primer 283 and oligo-dT11-G, A, and C primers, respectively. Dots denote unique PCR amplicons for the brain-passaged amoeba. G, A and C indicate oligo-dT primers used for PCR, the 3' terminal nucleotide of which was G, A and C, respectively. P, brain-passaged amoeba; N, non-brain-passaged amoeba; M, DNA size marker of 100 bp ladder.
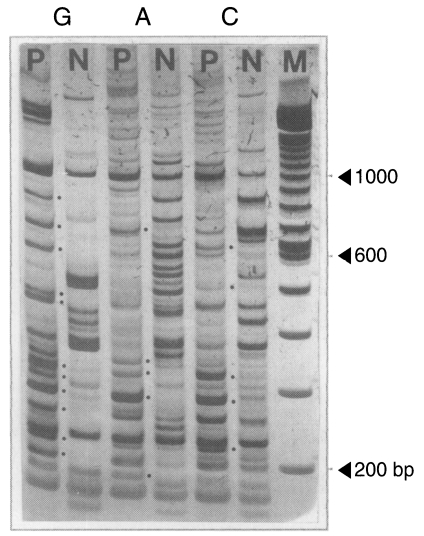
Fig. 2Identification of DDRT-PCR amplicons of differentially-expressed genes (arrow heads) by Southern slot-blot analysis using the pooled DDRT-PCR products as probes. PCR re-amplified products of the selected 96 unique amplicons for the brain-passaged amoeba were immobilized in duplicate sets onto nylon membranes and hybridized with the pooled original DDRT-PCR products from the brain-passaged amoeba (A) or from the non-brain-passaged amoeba (B).
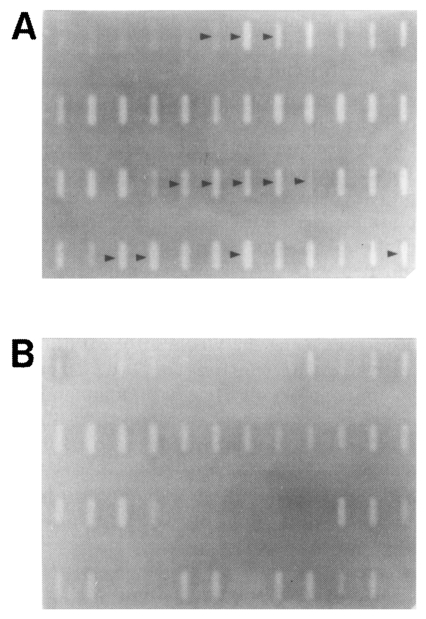
Fig. 3Nucleotide sequence and conceptual translation product of the clone A289C. The numbering refers to the nucleotide sequence and amino acid sequence.

Fig. 4Alignment of A289C with AmsG from Erwinia amylovora (EMBL accession No. Q46628) and cspD from S. agalactial (EMBL accession No. Q04664). Identical amino acids in two sequences are symbolized by bars. Conservative substitutions are marked by dots.

Fig. 5Upregulated expression of the A289C gene verified by Northern slot-blot analysis. 1, total RNA of brain-passaged Acanthamoeba culbertsoni; 2, negative control; 3, total RNA of non-passaged A. culbertsoni.

Table 1.Number of unique DDRT-PCR products observed only in brain-passaged Acanthamoeba culbertsoni
Table 1.
|
Primer |
dT-G |
dT-A |
dT-C |
Total |
|
235 |
5 |
5 |
NDa)
|
10 |
|
252 |
3 |
ND |
ND |
3 |
|
269 |
ND |
ND |
4 |
4 |
|
280 |
ND |
ND |
6 |
6 |
|
283 |
12 |
6 |
5 |
23 |
|
284 |
ND |
ND |
3 |
3 |
|
285 |
5 |
5 |
ND |
10 |
|
287 |
ND |
6 |
ND |
6 |
|
289 |
ND |
ND |
6 |
6 |
|
290 |
ND |
ND |
5 |
5 |
|
296 |
ND |
3 |
9 |
12 |
|
299 |
2 |
3 |
3 |
8 |
Table 2.DDRT-PCR products hybridized only with the probe synthesized from the pooled DDRT-PCR products of brain-passaged Acanthamoeba culbertsoni
Table 2.
|
Primer combinations |
Size (bp) |
|
dT-A : 235 |
310 |
|
dT-A : 235 |
420 |
|
dT-A : 287 |
170 |
|
dT-A : 296 |
220 |
|
dT-A : 296 |
300 |
|
dT-A : 299 |
460 |
|
dT-C : 289 |
300 |
|
dT-C : 289 |
440 |
|
dT-C : 289 |
460 |
|
dT-C : 289 |
550 |
|
dT-C : 289 |
750 |
|
dT-C : 299 |
650 |