Abstract
In order to evaluate the possible roles of secretory proteases in the pathogenesis of amoebic keratitis, we purified and characterized a serine protease secreted by Acanthamoeba lugdunensis KA/E2, isolated from a Korean keratitis patient. The ammonium sulfate-precipitated culture supernatant of the isolate was purified by sequential chromatography on CM-Sepharose, Sephacryl S-200, and mono Q-anion exchange column. The purified 33 kDa protease had a pH optimum of 8.5 and a temperature optimum of 55℃. Phenylmethylsulfonylfluoride and 4-(2-Aminoethyl)-benzenesulfonyl-fluoride, both serine protease specific inhibitors, inhibited almost completely the activity of the 33 kDa protease whereas other classes of inhibitors did not affect its activity. The 33 kDa enzyme degraded various extracellular matrix proteins and serum proteins. Our results strongly suggest that the 33 kDa serine protease secreted from this keratopathogenic Acanthamoeba play important roles in the pathogenesis of amoebic keratitis, such as in corneal tissue invasion, immune evasion and nutrient uptake.
-
Key words: serine protease, Acanthamoeba lugdunensis KA/E2, Korean corneal isolate, virulence factor
INTRODUCTION
Amphizoic amoebae of the genus
Acanthamoeba are ubiquitous in nature and are found in diverse habitats including soil, water, dust, air-conditioning units, domestic tap water, dental treatment units, contact lenses and lens cases (
Marciano-Cabral et al., 2000). In recent years,
Acanthamoeba species become a public health issue for two reasons (
Alfieri et al., 2000). First, the amoebae are recognized as the natural hosts for several intracellular microbes, which are pathogenic to humans, and second, several species of
Acanthamoeba have been implicated in human diseases, i.e., amoebic keratitis (AK) and granulomatous amoebic encephalitis (GAE). In contrast to GAE, which affects mainly immunocompromised hosts (
Sisson et al., 1995), AK affects young and healthy individuals particularly contact lens wearers (
Kilvington and White, 1994).
The mechanisms by which
Acanthamoeba invade host tissues are poorly understood. In AK, the adherence of trophozoites to corneal epithelial cells, which is presumed to be mediated by mannose recognition sites (
Yang et al., 1997;
Cao et al., 1998;
Leher et al., 1998) or a certain adhesion molecule (
Kennett et al., 1999), appears to be a critical step in the cascade of events, which leads to host tissue damage. Moreover, cytopathic factors, which lysed corneal epithelial cells in vitro, were found to be secreted into culture supernatant by trophozoites treated with mannose. And, the activity was almost completely inhibited by phenylmethylsulfonyl-fluoride (PMSF), a serine proteinase inhibitor (
Leher et al., 1998). This latter finding indicates that cytopathic effects can be mediated by a serine proteinase activity. Kong et al. (
2000) recently purified a 33-kDa secretory serine proteinase from
A. healyi, which was isolated from a GAE patient. The serine proteinase showed potent proteolytic activity upon various substrates such as type I and IV collagens, fibronectin, fibrinogen and hemoglobin, etc. By immunoscreening an
A. healyi expression library using antisera against the purified serine protease, Hong et al. (
2000) isolated a cDNA encoding a subtilisin-like serine proteinase (AhSUB1).
In the present study, we purified and characterized a 33 kDa proteinase secreted by Acanthamoeba lugdunensis KA/E2, which had been isolated originally from the infected cornea of a Korean AK patient. The enzymologic characteristics of this proteinase suggest that it may play significant roles in the pathogenesis of AK.
MATERIALS AND METHODS
Maintenance of Acanthamoeba lugdunensis KA/E2
Acanthamoeba lugdunensis KA/E2 was isolated from the infected cornea of a Korean keratitis patient, and was cultured axenically in a proteose peptone-yeast extract-glucose media at 26℃.
Assay of enzyme activity
Proteinase activity was assayed against casein. A mixture of 125 µl of 0.6% casein in 50 mM Tris (pH 8.5) and 20 µl of enzyme was incubated for 30 min at 55℃. The reaction was stopped by adding 125 µl of 0.4 M trichloroacetic acid and by incubating at 40℃ for 10 min. After centrifugation, 200 µl of the supernatant was mixed with 1 ml of 0.4 M sodium carbonate and 200 µl of Folin-Ciocalteu reagent and incubated at 40℃ for 20 min. The amount of degradation was determined by measuring absorbance at 660 nm. One unit of enzyme activity was defined as the amount of enzyme required to elaborate 1 µg of tyrosine per min from casein in a 1 ml reaction volume. The Bradford dye-binding procedure (
Bradford, 1976) was used to measure protein concentrations, except during chromatography where we used absorbance at 280 nm instead.
The amoeba culture supernatant was precipitated with 65% ammonium sulfate and centrifuged at 10,000 rpm for 15 min. The pellet was dissolved in 50 mM sodium acetate buffer (pH 5.2) and dialyzed against the same buffer at 4℃ for 30 hr. The dialyzed solution was then applied to a CM-Sepharose column (3.0 × 17 cm) equilibrated with 50 mM sodium acetate buffer (pH 5.2), and eluted with a 0-1 M NaCl gradient at a rate of 0.5 ml/min, with 5 ml of fraction volume. Each fraction was tested for enzymatic activity. The active fractions were pooled and dialyzed against 20 mM Tris-HCl (pH 8.8). The dialyzed solution was concentrated using a freeze dryer (Bondiro, Ilshine, Korea). The concentrated solution was loaded onto a Sephacryl S-200 column (HiPrep 1.6/60, Amersham, Sweden) and fractionated at a rate of 0.5 ml/min into 2.5 ml fraction volumes. Pooled active fractions were applied to a mono Q-anion-exchange column (Mono Q HR 5/5, Amersham, Sweden) and eluted with a 0-0.35 M NaCl gradient in 50 mM Tris (pH 8.0). Fractions showing proteinase activity were retained for further characterization.
Characterization of the purified enzyme
The molecular weight of the purified enzyme was determined by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and its proteolytic activity determined using casein as a substrate as described above. Its pH optimum was determined in sodium acetate buffer at pH 5.5-6.0, phosphate buffer at pH 6.0-8.0, and Tris-HCl buffer at pH 8.0-10.0 (Sigma Chemical Co). The temperature optimum of the purified enzyme was determined by incubating it at temperatures ranging from 4℃ to 65℃. To characterize the purified enzyme, phenylmethylsufonylfluoride (PMSF, 1 mM), diisopropyl fluorophosphate (DEF, 1 mM), L-transepoxysuccinyl-leucylamide (E-64, 10 µg/ml), dithiothreitol (DTT, 1 mM), pepstatin A (1 µg/ml), EDTA (1 mM) or phenathroline (1 mM) were preincubated with the enzyme for 20 min 37℃. Substrate was added and the proteolytic activity of the enzyme assayed. All chemicals were obtained from Roche Molecular Biochemicals (Mannheim, Germany), except for PMSF and DTT (Sigma, St. Louis, Missouri).
Substrate specificity
The substrate specificity of the purified serine proteinase was examined using 8 different substrates, namely, type I and type IV collagens, fibronectin, fibrinogen, hemoglobin, albumin, IgA and IgG. One hundred micrograms of each substrate was dissolved in 0.1 M sodium acetate buffer (type I and IV collagen) or 50 mM Tris-HCl pH 7.4 (the other substrates), to a concentration of 1 mg/ml, was mixed with 5 µl of the purified enzyme and incubated at 37℃. The concentration of the purified serine proteinase was 46 µg/ml for each reaction. Twenty microliters aliquots were withdrawn from the reaction mixture after 30 sec, 10 min and 30 min and electrophoresed in an acrylamide gel, after electrophoresis, the gels were stained with 0.1 % Coomassie blue.
RESULTS
Purification of proteinase
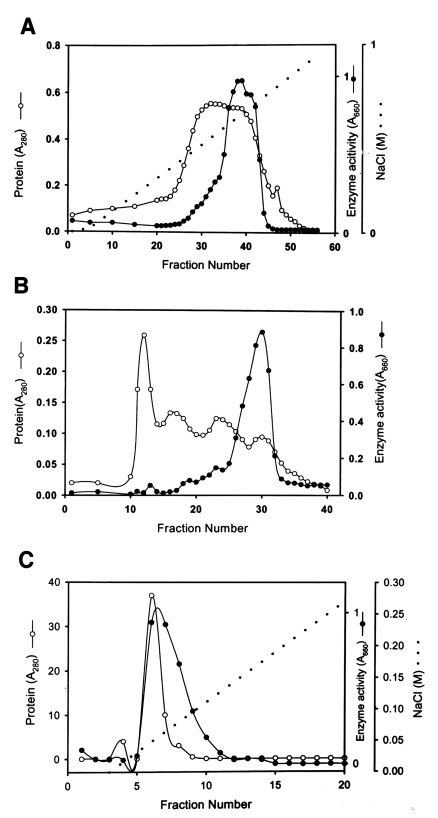
The purification used to separate the proteolytic enzyme from the culture supernatant of
Acanthamoeba lugdunensis KA/E2 are summarized in
Table 1 and
Fig. 1. A broad peak of enzymatic activity was eluted from CM-Sepharose between fraction numbers 33 and 43 at a NaCl concentration of 0.5-0.6 M (
Fig. 1A). Fraction numbers 36 to 42 were pooled and used for the further purification. A chromatogram of Sephacryl S-200 (
Fig. 1B) shows a major peak of activity between fraction numbers 26 and 32. Pooled active fractions (28-31) eluted from Sephacryl S-200 were applied to a mono Q-anion-exchange column.
Fig. 1C shows the peak protein concentration and proteolytic enzyme activity obtained from pooled fraction 6-7. Fraction 6 was used to characterize the substrate specificity of the enzyme. The specific activity of the purified enzyme was about 160-fold higher than that of the culture supernatant, and the enzyme was obtained at a yield of 3.2% (
Table 1).
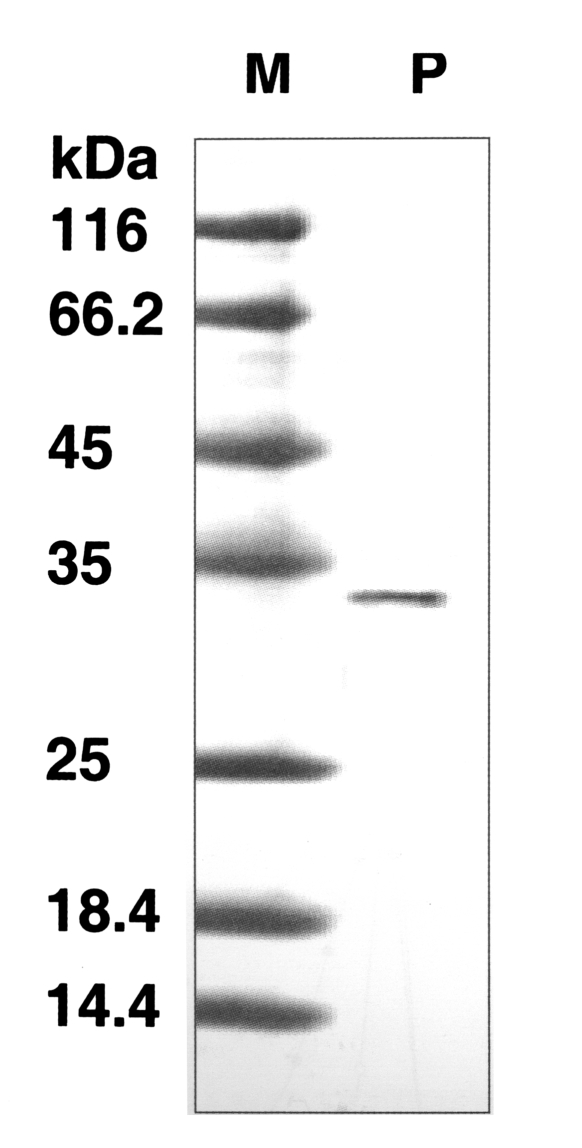
The molecular weight of the purified enzyme was about 33 kDa by SDS-PAGE (
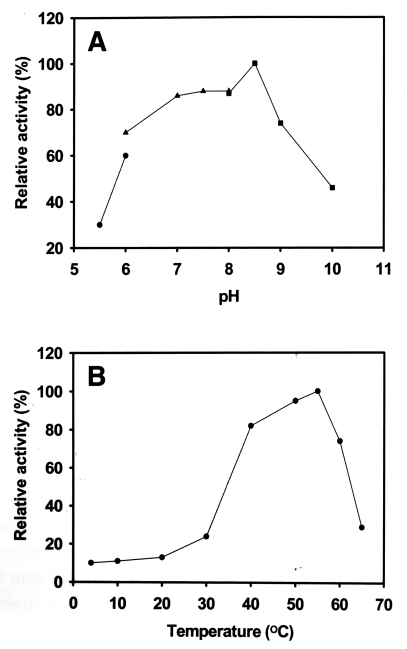
Fig. 2). The proteinase maintained its activity between pH 5.5-10.0, and showed maximum activity at pH 8.5 (
Fig. 3A) with an optimal temperature of 55℃ (
Fig. 3B). The activity of the enzyme was stable at 40-60℃, but its activity was ≤ 30% at 4-37℃. Serine proteinase-specific inhibitors, PMSF and DFP, completely inhibited the activity of the purified proteinase; other classes of inhibitor and activator had no effect on its activity (
Table 2).
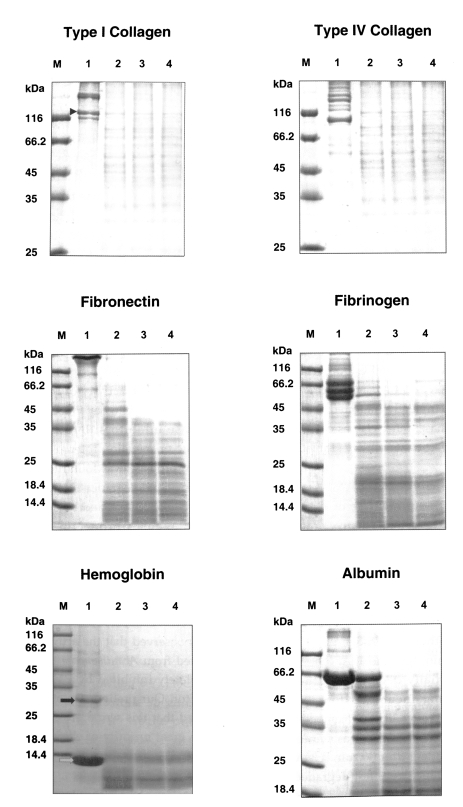
The purified serine proteinase of
A. lugdunensis KA/E2 degraded extracellular matrix protein, type I and IV collagens, and fibronectin into many small fragments (
Fig. 4). Moreover, the α-chains of type I collagen were cleaved into several small fragments (
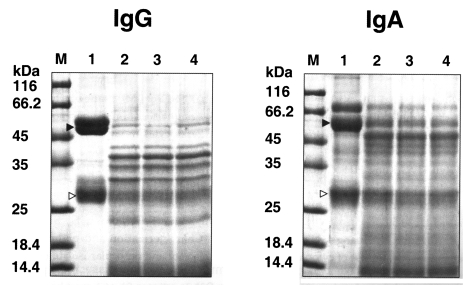
Fig. 4). The purified enzyme exhibited stronger activity against type I and IV collagens, and fibronectin than other substrates; i.e., fibrinogen, hemoglobin, albumin, IgG and IgA. Fibrinogen and albumin were partially degraded by the purified enzyme after being incubated for 30 sec, and completely degraded after 10 min. Hemoglobin dimer was degraded immediately but as monomer was incompletely degraded after 30 min. Heavy chains and light chains of IgA were not degraded after 30 min, however, the purified enzyme showed strong activity against the heavy chain of IgG and appeared weak activity against the light chain (
Fig. 5).
DISCUSSION
AK is a chronic progressive infection and does not resolve in the absence of effective chemotherapy. Moreover, failure to control the infection can lead to permanent loss of vision (
Ledee et al., 1996). In recent years, at least 20 patients have been diagnosed with AK in Korea (Hahn, personal communication). The most frequent type isolated from infected corneas and lens cases in Korea has been recognized as the L3a type (
Lee et al., 1997 and unpublished data) to which KA/E2 belongs. However, studies upon the virulence related factors, which are essential for the understanding of the pathogenesis of AK, have yet to be performed on Korean corneal isolates of
Acanthamoeba.
In the present study, we purified a secretory protease from the culture supernatant of Acanthamoeba lugdunensis KA/E2, which was isolated from the cornea of a Korean keratitis patient. Studies showed that proteolytic activity of this protease was almost completely inhibited by PMSF and AEBSF, both serine protease inhibitors, but unaffected by cysteine, aspartic or metallo-protease inhibitors. The purified 33 kDa protease was found to be active over a broad pH range and degraded many protein components of extracellular matrix and serum, including type I and IV collagens, fibronectin, fibrinogen, albumin, hemoglobin, IgG and IgA.
During the pathogenetic process of AK, amoebae may adhere to the corneal epithelial cells and secrete cytopathic or histolytic factors to invade the corneal stroma (
He et al., 1990;
Stopak et al., 1991). Leher et al. (
1998) observed that the activity of cytopathic factors secreted from
Acanthamoeba trophozoites was almost completely inhibited by PMSF, a serine proteinase inhibitor. Our preliminary study (unpublished) also showed that this serine protease activity was present in the great majority of clinical
Acanthamoeba isolates from 12 Korean AK patients. It is likely that like other tissue invading parasites,
Acanthamoeba employs secretory proteases to penetrate and feed upon the corneal stroma, and to evade the host's immunologic attack.
Type I collagen that make up over 90% of the corneal stroma is relatively resistant to degradation by the 33 kDa serine protease. Collagens are soluble at acidic pH but very insoluble at neutral or alkaline pH. The optimal pH for the protease purified in the present study was about 8.5. As mentioned above, AK has a chronic course. Therefore, in clinical infections, AK can efficiently degrade the type I collagen of corneal stroma.
IgA is the predominant immunoglobulin defense at the mucosal surface (
Underdown & Schiff, 1986). The biological functions of IgA are not completely understood, but include the ismmobilization and prevention of microbes adherence, the binding of toxins, and the inhibition of antigen absorption. The ability of the serine protease of
Acanthamoeba to degrade IgA may contribute, at least in part, to the evasion of the host's corneal immune response in AK.
We isolated a subtilisin gene product by PCR from Acanthamoeba lugdunensis KA/E2. Cloning of the full-length subtilisin gene and the expression of its recombinant enzyme may provide new informations on roles of this enzyme in AK. Future study will focus on the cytotoxicity and cytopathy of this purified protease on corneal cells.
Notes
-
This study was supported by a grant from the Korea Science and Engineering Foundation (#NO.RO5-2000-000-00105-0).
References
- 1. Alfieri SC, Correia CE, Motegi SA, Pral EM. Proteinase activities in total extracts and in medium conditioned by Acanthamoeba polyphaga trophozoites. J Parasitol 2000;86:220-227.
- 2. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248-254.
- 3. Cao Z, Jefferson DM, Panjwani N. Role of carbohydrate-mediated adherence in cytopathogenicmechanisms of Acanthamoeba. J Biol Chem 1998;273:15838-15845.
- 4. Hadas E, Mazur T. Biochemical markers of pathogenicity and virulence of Acanthamoeba sp. strains. Parasitol Res 1993;79:696-698.
- 5. He YG, Niederkorn JY, McCulley JP, et al. In vivo and in vitro collagenolytic activity of Acanthamoeba castellanii. Invest Ophthalmol Vis Sci 1990;31:2235-2240.
- 6. Hong YC, Kong HH, Ock MS, Kim IS, Chung DI. Isolation and characterization of a cDNA encoding a subtilisin-like serine proteinase (AhSUB) of Acanthamoeba healyi. Mol Biochem Parasitol 2000;111:441-446.
- 7. Kennett MJ, Hook RR Jr, Franklin CL, Riley LK. Acanthamoeba castellanii: characterization of an adhesion molecule. Exp Parasitol 1999;92:161-169.
- 8. Kilvington S, White DG. Acanthamoeba: Biology, ecology and human disease. Rev Med Microbiol 1994;5:12-20.
- 9. Kong HH, Kim TH, Chung DI. Purification and characterization of a secretory serine proteinase ofAcanthamoeba healyi isolated from GAE. J Parasitol 2000;86:12-17.
- 10. Ledee DR, Hay J, Byers TJ, Seal DV, Kirkness CM. Acanthamoeba griffini molecular characterization of anew corneal pathogen. Invest Ophthalmol Vis Sci 1996;37:544-550.
- 11. Lee SM, Choi YJ, Ryu HW, Kong HH, Chung DI. Species identification and molecular characterization of Acanthamoeba isolated from contact lens paraphernalia. Korean J Ophthalmol 1997;11:39-50.
- 12. Leher H, Silvany R, Alizadeh H, Huang J, Niederkorn JY. Mannose induces the release of cytopathic factors from Acanthamoeba castellanii. Infect Immun 1998;66:5-10.
- 13. Marciano-Cabral F, Puffenbarger R, Cabral GA. The increasing importance of Acanthamoeba infections. J Eukaryot Microbiol 2000;47:29-36.
- 14. Mitro K, Bhagavathiammai A, Zhou OM, et al. Partial characterization of the proteolytic secretions of Acanthamoeba polyphaga. Exp Parasitol 1994;78:377-385.
- 15. Sison JP, Kemper CA, Loveless M, McShane D, Visvesvara GS, Deresinski SC. Disseminated Acanthamoebainfections in patients with AIDS. Clin Infect Dis 1995;20:1207-1216.
- 16. Stopak SS, Roat MI, Nauheim RC, et al. Growth of Acanthamoeba on human corneal epithelial cells andkeratocytes in vitro. Invest Ophthalmol Vis Sci 1991;32:354-359.
- 17. Underdown BJ, Schiff JM. Immunoglobulin A: Strategic defense initiative at the mucosal surface. Annu Rev Immunol 1986;4:389-417.
- 18. Van Klink F, Alizadeh H, Stewart GL, et al. Characterization and pathogenic potential of a soil isolate and an ocular isolate of Acanthamoeba castellanii in relation to Acanthamoeba keratitis. Curr Eye Res 1992;11:1207-1220.
- 19. Yang Z, Cao Z, Panjwani N. Pathogenesis of Acanthamoeba keratitis: carbohydrate-mediated hostparasite interactions. Infect Immun 1997;65:439-445.
Fig. 1Chromatograms of proteinase purification from the culture supernatant of Acanthamoeba lugdunensis KA/E2. A. CM-Sepharose chromatography of the ammonium sulfate-precipitated culture supernatant. B. Chromatogram of Sephacryl S-200 column of the enzymatically active fractions (fractions 36-42) from A. C. Chromatogram of enzymatically active fractions (fraction numbers 28-31) of B from a mono Q anion-exchange column.

Fig. 2SDS-PAGE of the purified enzyme in a gradient acrylamide gel. A single protein band (arrow) stained with Coomassie blue was observed with a size of approximately 33 kDa. Lane: M, standard size marker: P, purified proteinase.

Fig. 3Effect of pH and temperature on serine proteinase activity. A. The enzyme activity was assayed in sodium acetate (pH 5.5-6.0), phosphate (pH 6.0-8.0), or Tris-HCl (pH 8.0-10.0). B. Optimum temperature of the enzyme. Enzyme activity is expressed as a percentage of the maximum proteolytic activity observed.

Fig. 4Substrate specificity of the purified secretory serine proteinase of A. lugdunensis KA/E2. M, protein size standard; Lane 1, reaction mixture without enzyme; 2-4, incubation for 30 sec, 10 min, 30 min; M, protein size standard. Marks indicate α-chains of type I collagen (▶) monomers (➡) and dimers (⇨) of hemoglobin.

Fig. 5Degradation of IgG and IgA by the serine proteinase of A. lugdunensis KA/E2. Lane 1, reaction mixture without enzyme; 2-4, incubation for 30 sec, 10 min, 30 min; M, protein size. The heavy (▶) and light chains (▷) of immunoglobulins are indicated.

Table 1.Purification steps of the serine proteinase obtained from the culture supernatant of Acanthamoeba lugdunensis KA/E2
Table 1.
|
Step |
Total protein (mg) |
Total activity (μg/min) |
Specific activity (μg/min/mg) |
Purification (fold) |
Yield (%) |
|
Culture supernatant |
156.3 |
813.0 |
5.2 |
1.0 |
100.0 |
|
Ammonium sulfate precipitation |
28 |
770.0 |
27.5 |
5.3 |
94.7 |
|
CM-Sepharose |
3.65 |
243.1 |
66.6 |
12.8 |
29.9 |
|
Sephacryl S-200 |
0.25 |
142.6 |
570.3 |
109.7 |
17.5 |
|
Q-anion exchange |
0.03 |
26.1 |
814.4 |
156.6 |
3.2 |
Table 2.Effects of enzyme inhibitors and activators on the activity of the purified proteinase
Table 2.
|
Inhibitor/activator |
Final Concentration |
Activity as percent of control |
|
Control |
|
100.0 |
|
Serine class |
|
|
|
PMSF |
1 mM |
6.9 |
|
DFP |
1 mM |
15.9 |
|
Cysteine class |
|
|
|
E-64 |
10 μg/ml |
96.7 |
|
DTT |
1 mM |
85.5 |
|
Aspartic class |
|
|
|
Pepstatin A |
1 μg/ml |
97.4 |
|
Metallo class |
|
|
|
EDTA |
1 mM |
108.3 |
|
1, 10-Phenanthroline |
1 mM |
107.2 |